

AI-ACCELERATED DRUG DISCOVERY
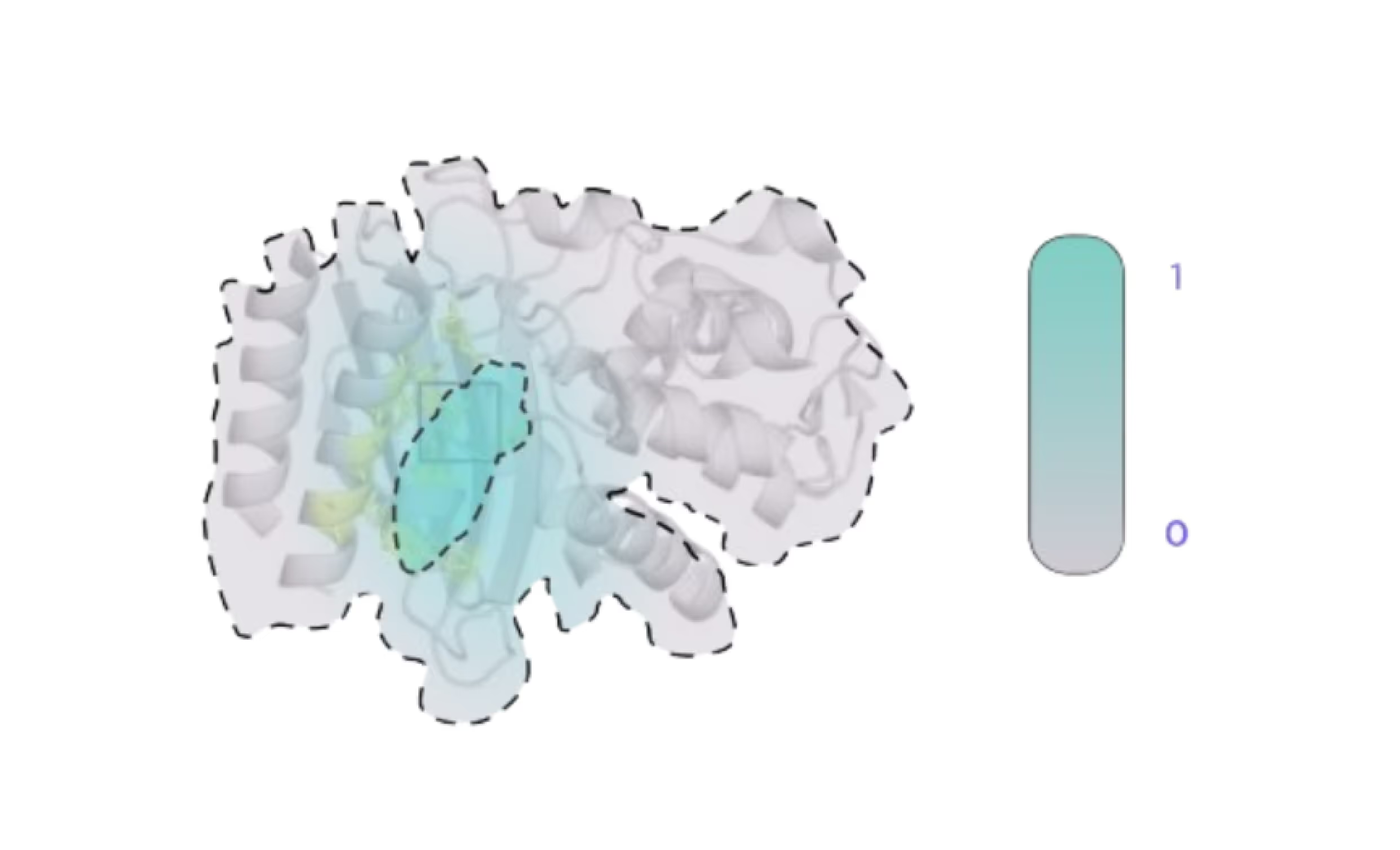
In the first “bootstrapping” stage the conformational changes in the target protein are induced to reveal the cryptic pockets and make them accessible to the drugs.

These molecules could either bind to the cryptic pockets themselves stabilizing them in open conformation or bind to the remote allosteric sites which induce corresponding changes in protein conformation or dynamics. Bootstrapping binders are chosen from the stock chemical spaces or readily available fragment libraries, which makes them cheap and fast to deliver.
In the second stage the protein complexes with the best predicted bootstrapping binders are assessed by molecular simulations and the high throughput structure determination (CryoEM).


This allows to elucidate the conformational changes, induced by the binders and to generate an ensemble of representative protein conformations, which contain open and accessible cryptic pockets. This ensemble is then passed through AI-based pocket prediction and tentative cryptic pockets are identified and annotated.
The production virtual screening using the full power of the Receptor.AI ecosystem is performed against each of the tentative pockets and the candidate compounds are selected from the large and diverse combinatorial chemical space.


The third stage starts from validation of the candidate compounds’ biological activity. The protein complexes with the active compounds are subject to another round of structure determination to confirm their binding to predicted cryptic pockets and to obtain additional data about binding mechanisms.
Confirmed compounds are designated as hits and are passed to the lead discovery and lead optimization stages using the fit-for-target Receptor.AI workflow.
