The chemical features which make drugs selective to several targets at once could be deduced from predictive AI models
AI contributes to understanding polypharmacology with explainable ML
The chemical features which make drugs selective to several targets at once could be deduced from predictive AI models
Announcement


Full Text
Modern pharmacology is dominated by the “strict selectivity” paradigm. The drug is usually considered “good” if it interacts selectively with a predefined molecular target, associated with a particular disease or condition. Such an approach is stimulated by the gene-centered view where the protein produced by the proven disease-associated gene is considered as a sole target.
This somewhat naive approach, which neglects complex molecular interactions and multiple intersecting metabolic and signaling pathways in cells, is already considered outdated. Nowadays successful drugs should block particular pathways of cellular processes rather than individual proteins. This inevitably leads to the polypharmacology approach: the ideal drug should act on multiple molecular targets associated with malfunctioning cellular networking. This ensures that there are no unexpected “sideways” which allow the disease to bypass the treatment, which is especially important for cancer and antibacterial treatments.
However, polypharmacology is much more complex than targeting a single protein. It requires a much better understanding of the structure-activity relationship of the drugs, because the same molecule binds to several unrelated targets and causes different effects. Moreover, there is an intuitive feeling among experts in medicinal chemistry that strictly selective compounds are fundamentally different from those, which bind to several targets. However, this intuition is very hard to rationalize and to translate into formal rules.
When speaking about distinguishing between something vaguely defined and poorly perceived by human intelligence, the AI classifiers usually come to rescue. There is little doubt that AI can be trained to distinguish single-target and multiple-target drugs. The problem is that ordinary AI models are opaque black boxes, which gives us no clue on how exactly they make a decision. If we, humans, want to understand what makes certain chemical structures multiple specificities, we need an explainable AI, which explicitly rationalizes the features in data leading to particular classification result.
The recent paper in Nature Scientific Reports is devoted to building such an explainable AI model, which reveals the distinctive features of dual-target and single-target compounds. This is only a subset of the more general task of revealing polypharmacology features, but it demonstrates how AI predictions could be translated back to human-comprehensible chemical knowledge.
The authors used three unrelated targets: monoamine oxidase B (MAOB), the A2a adenosine receptor (A2aR), and acetylcholinesterase (AChE), which are all popular protein targets related to the central nervous system diseases. MAOB was chosen as a target, which is shared by all dual-target drugs.
The dataset contained 52 double-target ligands for the MAOB-A2aR target pair and 70 for the MAOB-AChE pair. The number of single-target ligands was larger and constitute 1932 / 3566 for the first target pair and 1793 /1853 for the second pair. Different number compounds for MAOB in both pairs is explained by the fact that not all of them are annotated with additional reported activities A2aR or AChE.
For each target pair, the balanced random forest classification models were constructed to distinguish between double-target and single-target drugs. For accurately predicted compounds, specific representation features were identified that were responsible for classifying them into single or double-target group. These features were then mapped back onto compounds to examine whether they formed meaningful chemical substructures, leading to the detection of characteristic structural motifs. The Shapley Value analysis was performed to determine the relative importance of different model features on prediction.
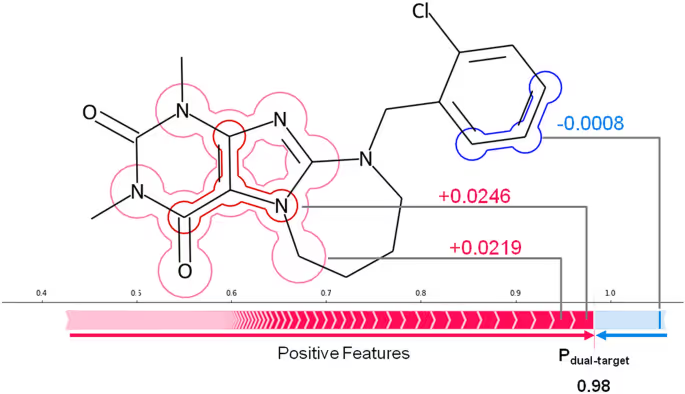
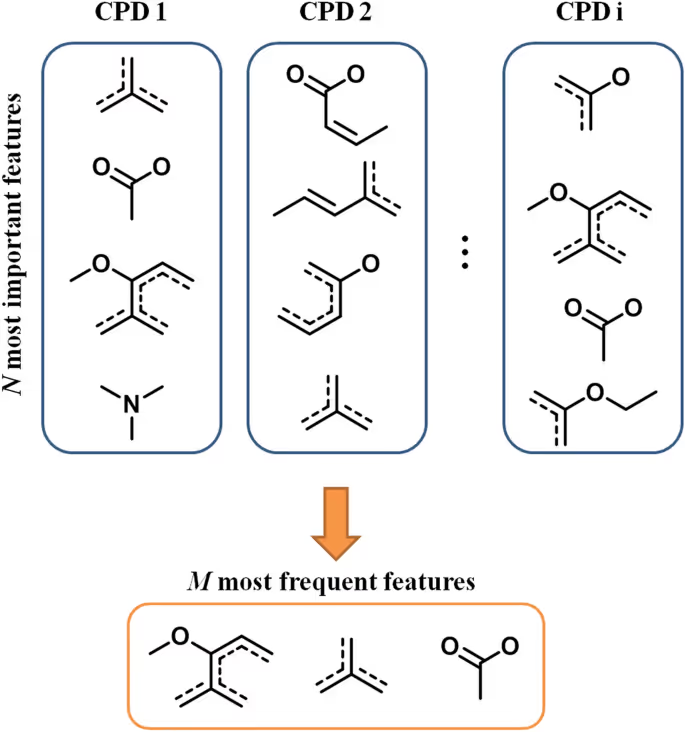
In order to extract the features in a reliable and robust way, the ranked scheme was used. On the basis of SV values, the most important features present incorrectly predicted double-targeted compounds were selected. After that given number of features occurring most frequently across all these compounds were prioritized.
After that the features were mapped onto the actual chemical structures. Individual atoms were assigned a weight equal to the number of important features containing them. This allows creating a “heat map” of chemical groups importance for double-targeting the compound.
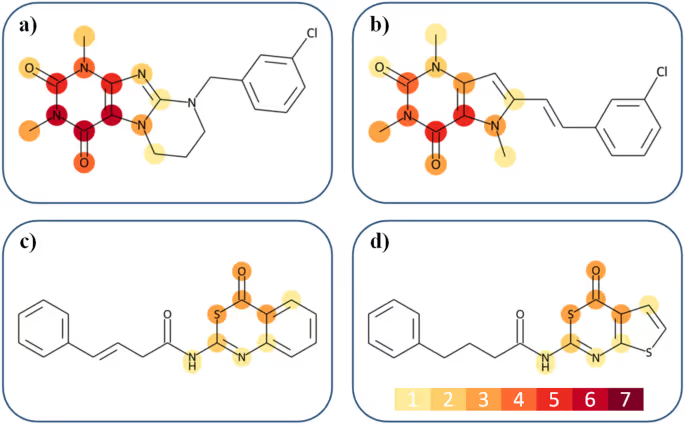
The results of our study show the capabilities of explainable ML to combine robust predictions of single or double-target activity with human-comprehensible identification of chemically relevant characteristics of the molecules. The established scheme is applicable to other target combinations and compound features.
Such analysis is useful not only for human comprehension but also for seamless incorporation into the AI-based drug discovery pipeline, namely the hit-to-lead and lead optimization stages. If the atoms and chemical groups of the hit compounds, which are responsible for polypharmacological effects, are known then such groups could be included into the desirable molecular scaffold while other parts of the molecules could be subject to changes in the lead series. In contrast, if any off-target interactions should be avoided, the opposite optimization strategy could be applied.
Receptor.AI is paying special attention to polypharmacology and currently implements multiple techniques for testing an off-target activity and boosting lead specificity to one or several desired proteins.