AI Drug Discovery: Not One-Size-Fits-All – Addressing Data Challenges
Announcement


Full Text
In the last 2 years, most of us have started using AI tools on an everyday basis. It seems the AI revolution came from nowhere, expanding its influence on all aspects of our lives and all industries. However, there is a field where the AI revolution started 10 years ago - Drug Discovery.
Nowadays, almost every big pharma company has already integrated some AI workflows into their drug development pipelines. Furthermore, you will struggle to impress anyone from the industry with yet another AI solution, pretending to minimize experimental work in drug discovery - this is already taken for granted.
Does it mean that we are closer than ever to an “ultimate drug discovery AI”, which will make all current tools and platforms obsolete? Are we going to witness a success comparable to ChatGPT in Pharma soon? For better or worse, such a breakthrough is very unlikely to happen in the foreseeable future. In this article, we discuss the major fundamental issue in AI-aided drug design - data scarcity, and how it changes the field and approaches used.
Training data issue
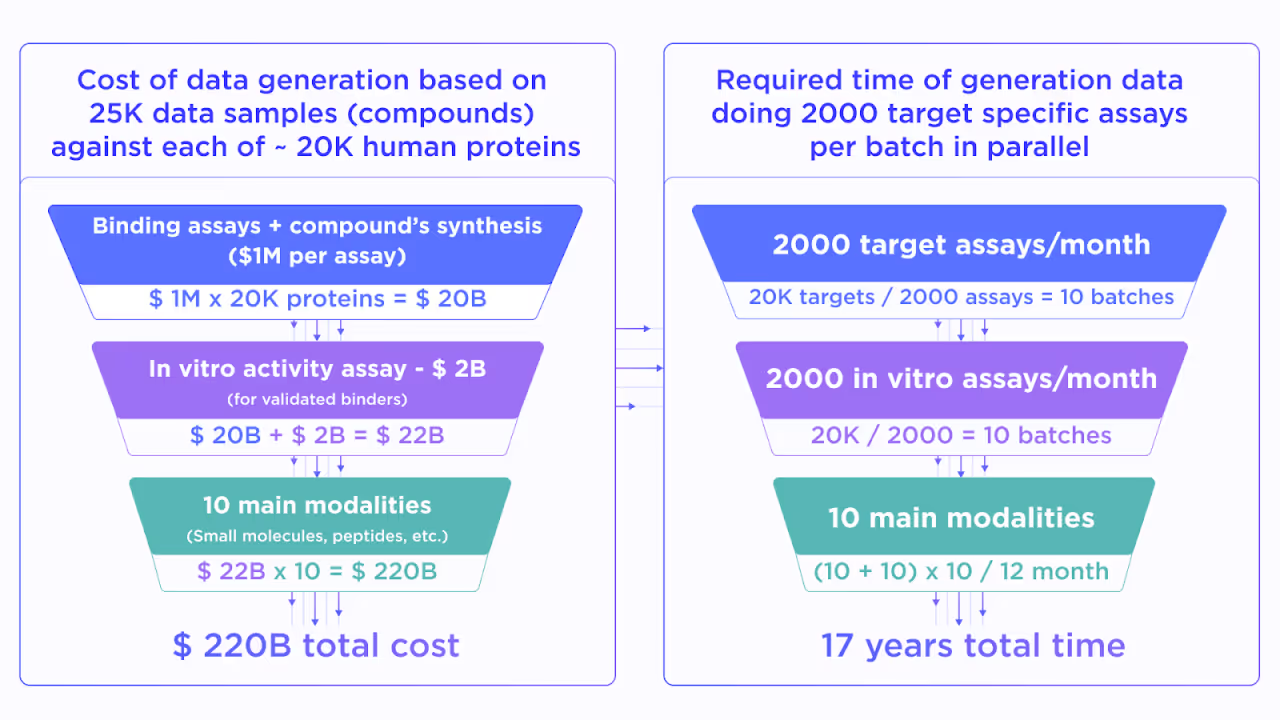
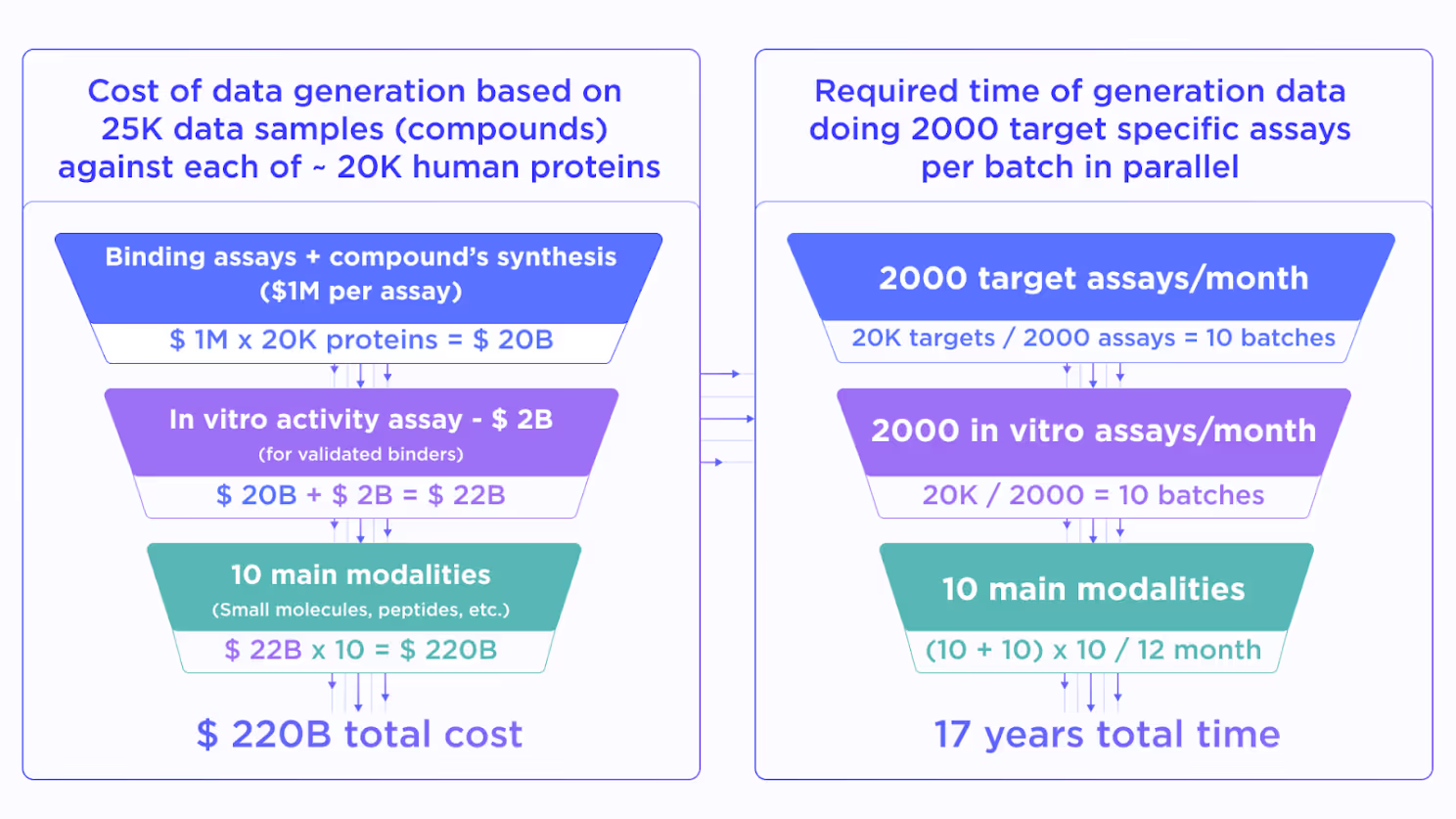
Justifying the previous statement, let us imagine the project which aims to build an “ultimate drug discovery model”. Such a model should be able to generate the molecules, which are suitable for IND enabling and clinical studies from the first shot, without going through the usual lengthy and expensive design-make-test iterative optimization process. Of course, the model has to be general enough to work with any of about ~20K potential target proteins in the human proteome.
To build such a model one needs comprehensive data for binding and in vitro activity of a diverse set of compounds. Let us assume that 25K molecules cover enough variability in the drug-like chemical space. This is a very optimistic estimate due to the billions of compounds currently available in virtual chemical spaces.
Then, we approximate the cost of a high-quality in vitro binding assay for 25K compounds as $1M. It must be performed for ~20K human proteins to get the proteome-wide training data coverage for the target-agnostic AI model. This would cost about ~$20B. On top of this, we have to perform an in vitro activity assessment for all validated binders which adds around $2B to the total price.
Resulting in $22B quite a lot, but still not totally unfeasible for the pharmaceutical industry. However, we still didn’t take into account the existence of multiple drug modalities, such as non-covalent and covalent small molecules, macrocycles, linear and cyclic peptides, peptidomimetics, bispecific compounds, antibodies, various conjugates, etc. This forces us to multiply the already stellar price by a factor of 10 reaching nearly $220B.
While being absolutely massive such an investment still looks reasonable if the resulting AI will substitute all preclinical drug discovery worldwide. However, what makes it unreal is the required time. Even with resources for ~2000 assays for all needed modalities together with properly expressing the protein of interest, we will spend approximately 17 years on 20K proteins, which obviously won’t solve the present-day struggles in drug design.
Lowering fidelity
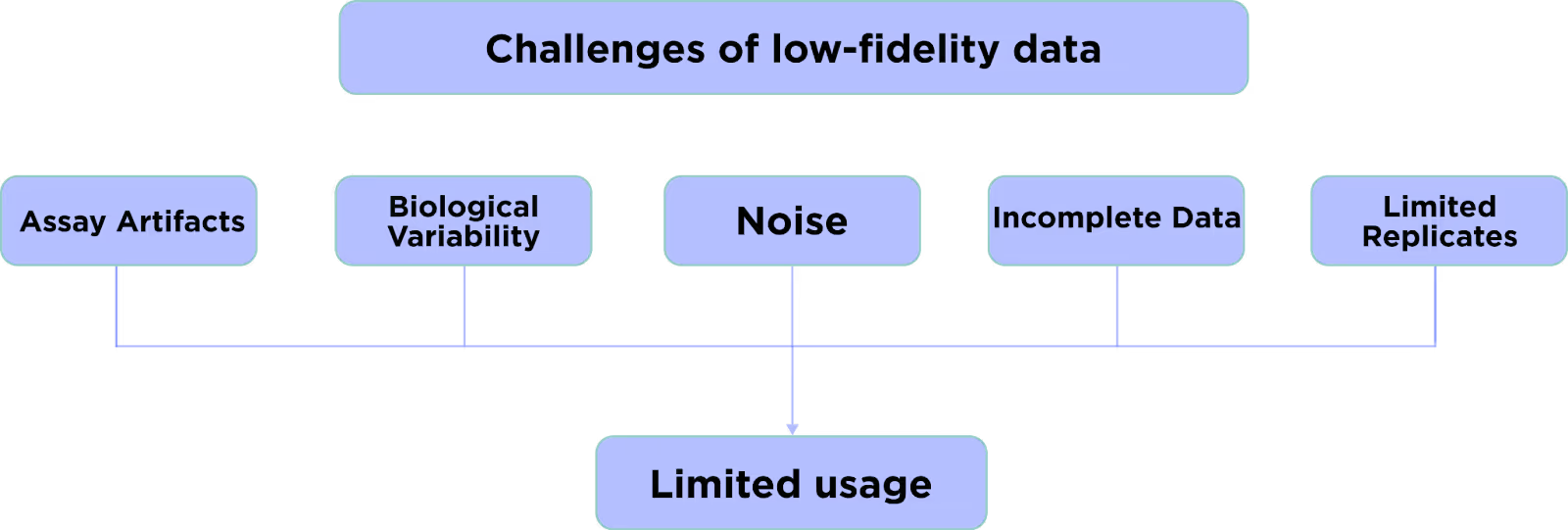
Therefore, it is clear that we cannot brute-force the data scarcity challenge just by performing the needed amount of experiments. However, what if we consider the data that already exists? The thousands of companies and academic institutions constantly generate results from various assays across different targets and modalities. The main issue with such data is its quality.
Most of the early-stage data in drug discovery have low fidelity i.e. significant noise, systematic biases, high variance, many false positives and negatives, and other deficiencies. The AI models trained on such unreliable data will inevitably give unreliable results.
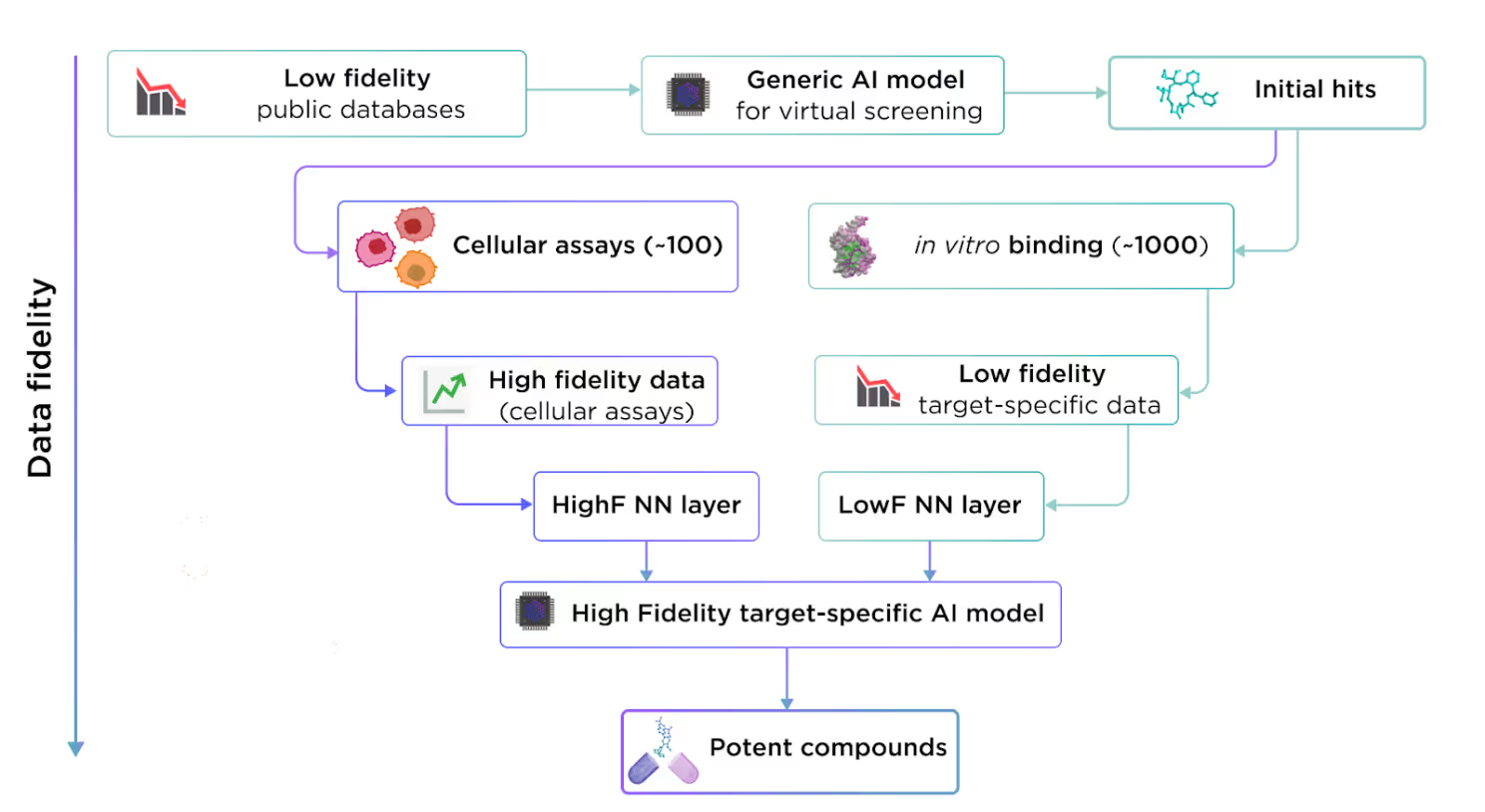
Still, there is a good way to employ low-fidelity data in AI drug design. Despite the fact it cannot be used for training the high-quality production model, it still covers a lot of variability which is otherwise not possible to cover with an inherently limited amount of high-fidelity data. Here is the place where transfer learning comes into play.
Transfer learning
The main idea of transfer learning is to fine-tune the generic pre-trained model with more case-specific data. The classic example is a face-recognition model, which is used in Face ID and similar technologies. The generalized model is pre-trained to discriminate faces from anything else and the fine-tuning trains it to recognize the face of a particular person.
This concept is highly applicable to drug design, where we can leverage extensive public low-fidelity data to train a generalized model. This generalized model can then be fine-tuned with high-fidelity target-specific data to address particular drug targets.
Instead of testing experimentally ~20K proteins on tens of thousands of compounds, we can test a smaller batch of prospective compounds on one protein of interest. The data obtained from these experiments will be sufficient to fine-tune a target-specific model, capable of producing high-quality results for that particular project.
This approach is less ambitious than creating an ultimate universal drug design model, but it is technically feasible and works in real-world scenarios now without the need to invest hundreds of billions over many years.
Data augmentation
Transfer learning is great but it won’t save you if available low-fidelity data is not enough to achieve the decent performance of the generalized model. An example of such a problem is AI docking. There are only about 55K resolved protein-ligand complexes, which form a highly redundant and skewed dataset, severely biased towards well-studied proteins and widespread classes of ligands. One can tackle these problems with very complex and sophisticated ML architectures, which can take the most of the available 55K complexes, but the price would be very slow in inference speed and the risk of overfitting.
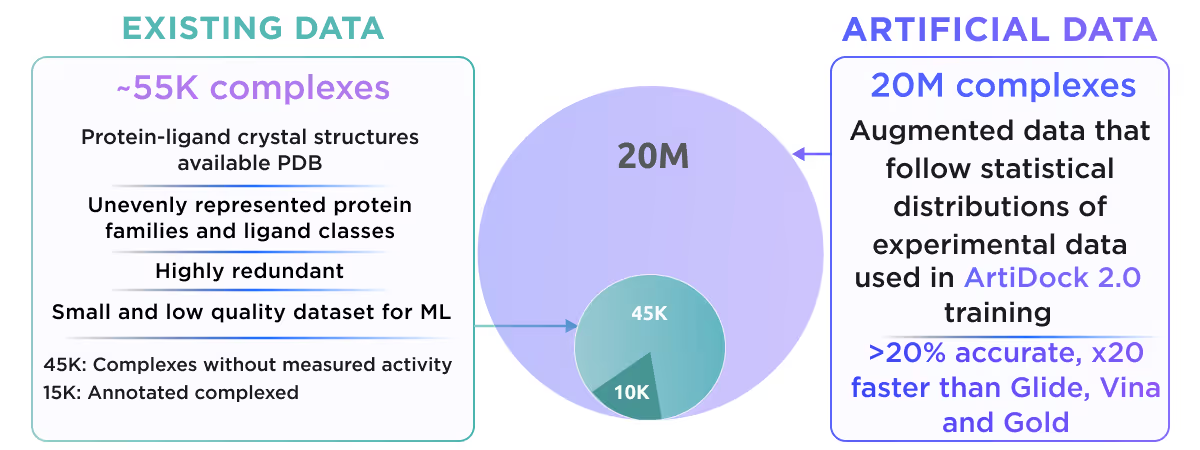
We at Receptor.AI decided to follow a different way. We deliberately chose simple and fast ML architecture and employed a multimodal data augmentation to overcome the shortage of data. Our approach is based on two augmentation techniques - generation of numerous artificial binding pockets around diverse ligands and sampling of the dynamics of about ~17K existing complexes with molecular dynamics simulations. Using this strategy we increased the initial dataset by ~1000 fold, which gives us a huge boost in model accuracy and robustness.
This resulted in the creation of ArtiDock - an AI docking model with accuracy comparable to AlphaFold 3, but 600x faster. You can read more about ArtiDock in our preprint.
Conclusions
AI has been a part of drug discovery for over a decade, but creating an "ultimate drug discovery AI" is still out of reach due to data scarcity and data quality issues. The cost and time required to collect enough data for such a “killer model” is currently beyond the reach of big pharma and tech giant companies alike.
However, techniques like transfer learning and data augmentation provide viable practical solutions. By fine-tuning generalized models with target-specific data and by expanding limited datasets through data augmentation, we can develop high-quality focused AI models that vastly speed up drug discovery projects. These methods, while not providing a perfect general-purpose solution, greatly enhance the efficiency and accuracy of AI-driven drug development. We envision the trend in the creation of specialized solutions for training AI models, tailored to specific targets and modalities, on the fly for each individual drug discovery project.