AI vs Physics: Present and Future of PPI Structure Prediction Methods
Announcement


Full Text
Understanding Protein Interactions: Key to Disease Treatment
Proteins are fundamental components that manage vital cellular functions. They scaffold sub-cellular structures, catalyze biochemical reactions, and facilitate adaptation to the environment. Central to these functions are protein-protein interactions (PPIs). By understanding these interactions, we can unravel the mechanisms underlying pathologies 1.
Techniques like co-immunoprecipitation, TAP-MS, and protein microarrays help identify interacting proteins within disease contexts. However, these top-down approaches often lack the atomic-level detail essential for verifying interaction mechanisms and guiding rational drug design 2.
Wet lab approaches to determine PPI complex structure
Considering the importance of PPIs, advanced experimental techniques have been developed to determine complex structures at the molecular level. Methods like cross-linking mass spectrometry (XLMS) and nuclear magnetic resonance (NMR) can rapidly provide insights within days but are limited by resolution and system size (< 1K atoms). Conversely, X-ray crystallography and cryo-electron microscopy (Cryo-EM) are not limited by the system size but demand significantly more time, ranging from weeks to months or even years 3.
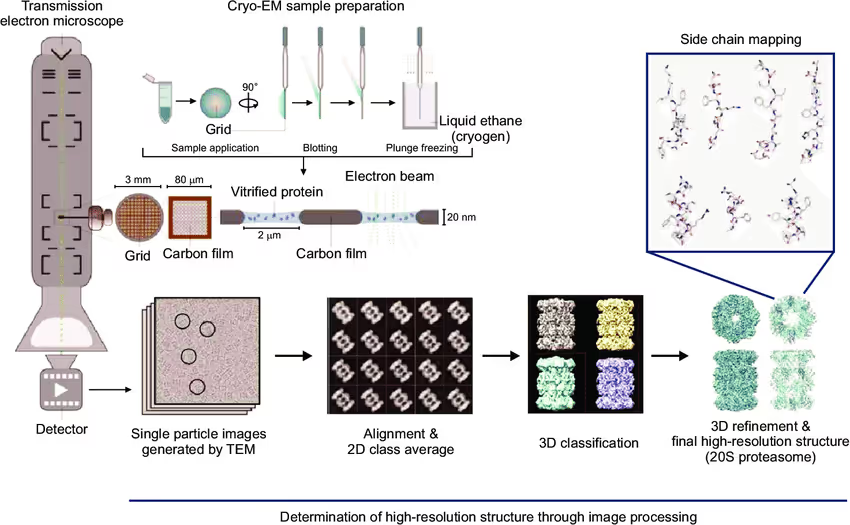
X-ray crystallography is limited to PPI targets that can be crystallized, while Cryo-EM offers greater flexibility by capturing transient interactions and conformational shifts. Despite these advantages, the low throughput of Cryo-EM still limits its wide application, driving the demand for alternative approaches.
Computational Predictions: Speed and Scalability
The time constraints of wet lab approaches and the complexity of biological systems prompted the development of computational approaches. The first methods that addressed the PPI prediction problem were physics-based. They are mostly represented by protein-protein docking algorithms (PPDAs) 4.
PPDAs mainly rely on sampling random rotations and translations of the structures of interacting proteins in order to find the most favorable orientation. The resulting complexes are ranked by score based on geometric complementarity and interaction energy.
Advanced PPDAs increased in accuracy by introducing the conformational sampling stage: exploring the flexibility of the backbone and sidechains (i.e., using statistical-derived backbone and rotamer libraries or lightweight molecular dynamics simulations routines) and/or the dynamic scoring approaches 5.
The AI Advantage in Structural Predictions
In the last few years, AI structure prediction methods (including those for PPIs) have surged dramatically. They are trained on protein structures stored in public repositories (i.e., PDB) and have three foundational elements 6:
- Encoding protein sequences into the representations that capture key residue interactions, whether derived from PLMs, MSAs, or latent embeddings.
- Conversion of these representations into accurate full-atom 3D models by integrating local context and long-range spatial relationships.
- Progressive refinement of predicted structures through iterative optimization guided by physical constraints.
Direct protein structure prediction reduces atomic clashes and suboptimal geometry typical in rigid docking methods. In an earlier benchmark, traditional docking methods predicted fewer than 25% of protein interactions accurately, while modern state-of-the-art AI methods achieved about 45% accuracy (Docking Benchmark Set 5.5) 7.
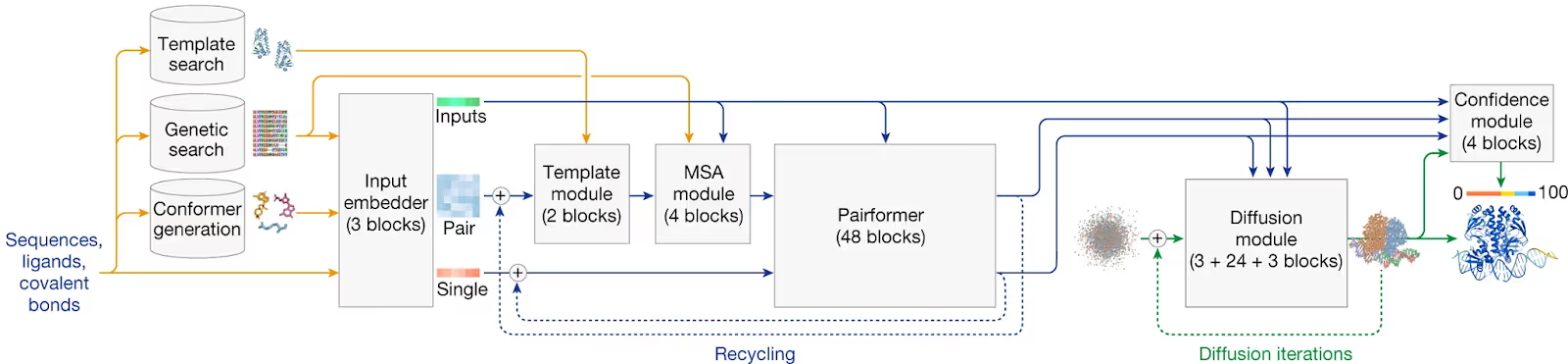
The most recent state-of-the-art AI-based methods for PPI structure prediction are AlphaFold-Multimer (AFM), LatentDock (LD), AlphaFold3 (AF3), and DFMDock. These methods, trained on similar datasets, differ in their architectural approaches:
- AFM (2021) is an adaptation of AlphaFold2, fine-tuned for PPI prediction. It leverages MSAs to detect co-evolutionary signals, facilitating accurate mapping of PPI contacts.
- LD (2023) utilizes latent representations derived from structural data and employs a denoising diffusion model for sampling amino acid pairings. This approach enhances inference speed with minimal accuracy loss.
- DFMDock (2023) employs a diffusion model with roto-translational equivariant representations of amino acids, treating them as graph nodes. Its integration of energy-based scoring directly into structure sampling reduces the need for separate confidence models.
- AF3 (2024) introduces a diffusion module, replacing the previous structure generation module, and expands its predictive capabilities to include interactions with DNA, RNA, and small molecules. This architectural change aims to improve accuracy, especially for antibody-antigen interactions.
AI Methods: Promising but Imperfect
Despite the great progress of AI-based techniques for PP structure prediction in terms of accuracy and/or efficiency optimization, there are a number of drawbacks to be addressed.
- Imbalance Between Positive and Negative Samples: In PPI datasets, known interactions (positive samples) are often much fewer than non-interactions (negative samples). This imbalance biases AI models, affecting their accuracy.
- Limited utility of confidence metrics: Scores like ipTM and ipAE accurately reflect structural quality but inadequately represent interaction strength or evaluate alternative interaction modes.
- Template and evolutionary data dependence: Improved predictions from structural templates are valuable but often unavailable if there are no homologous structures resolved. Thus, PPIs lacking evolutionary or template information remain challenging for current methods.
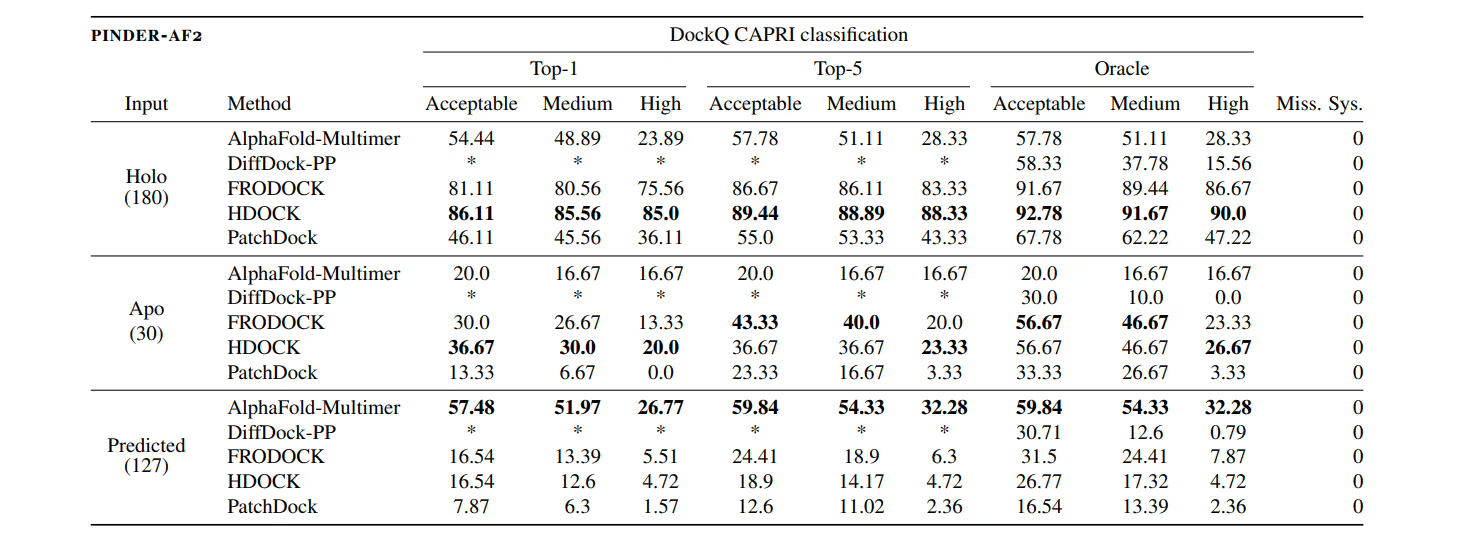
In the recently released PINDER benchmark, AlphaFold-Multimer performed worse than the physics-based methods in most cases, with a 2 times lower accuracy in the number of tests. This reveals the suboptimal performance of AI methods that block their effective usage in drug design 8.
Injecting Physics into AI at Receptor.AI
So far, the community has mainly focused on optimizing AI-based methods by leveraging more efficient inference pipelines or optimized MSA pre-processing steps 9. However, the accuracy under the lack of structural templates and evolutional information still lags behind the physics-based techniques.
At Receptor.AI, we suggest that embedding physical knowledge into AI PPI prediction methods could allow them to surpass these limitations. We are investigating several ways to integrate physics-based approaches with the inference of internal and 3rd-party SOTA PPI prediction models:
- AI conformational sampling and PPDA integration.
- Optimization of our AI-based PPI pattern detection and protein complex assembly with further physics-based optimization.
- Several other approaches will be disclosed later.
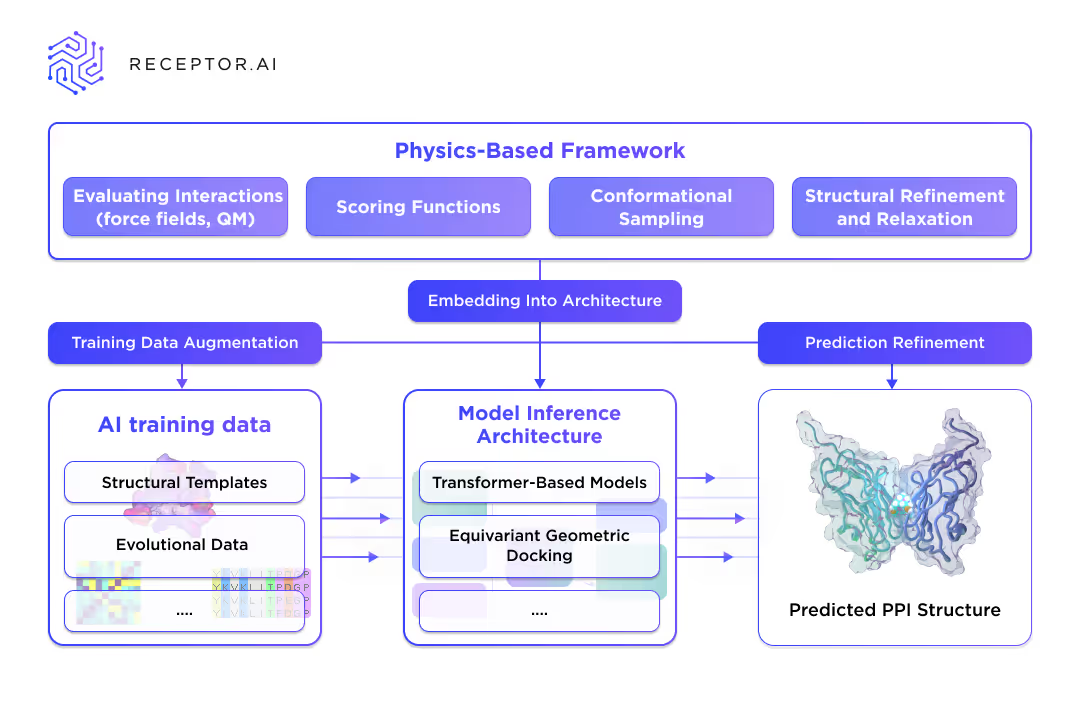
The preliminary data demonstrate the potential for significant improvement in challenging cases with which current methods struggle.
Furthermore, as our research progresses, we look forward to sharing our comprehensive findings. These forthcoming data will provide deeper insights into the benefits and limitations of integrating physics-based elements with AI-driven PPI prediction.