Benchmark of the AI-augmented structure-based activity and affinity prediction methods combined by a smart consensus function
Announcement


Full Text
Structure-based techniques for affinity and activity prediction are of great importance at all steps of virtual screening, but especially in the later stages where primary filtering of the large chemical space has already been performed by faster AI methods. These techniques allow for a more granular evaluation of the shortlisted compounds, which explicitly takes into account the target protein structure in general and the architecture of its binding pocket in particular. Structure-based techniques are, on average, more computationally demanding, thus, their application is justified at the secondary screening stage.
Receptor.AI developed several techniques used for the structure-based virtual screening: the drug-target interaction model (DTI), the fragment-based drug-target interaction model (FB-DTI) and the custom docking with AI rescoring, which evaluates compounds’ activity based on the docking poses.
We have also tested a combination of several techniques, which contribute to a smart consensus function and compared them with the set of experimentally determined activities for several target proteins. The automated optimisation procedure allows us to choose the best functional form of consensus function and its parameters from a broad space of possible variants.
Here we present the benchmarks of these techniques.
The overview of performed tests
- DTI benchmark evaluates the model performance on a dataset containing pairs of known active compounds with their corresponding target proteins from the public databases.
- FB-DTI benchmark evaluates the model performance on a dataset containing pairs of molecular fragments and protein active site subpockets from known protein-ligand complexes in PDB.
- Docking Rescoring benchmark. The model was trained on the set of all known crystal structures of the proteins with their active ligands and tested on the subset of this data, which was excluded from the training set.
- Comparison to the brute-force docking. Our AI-based techniques were compared to 16 most widespread docking algorithms on the high-quality manually curated testing dataset of 8 commonly used target proteins with their specific ligands.
- Benchmarking of the smart consensus function, which combined DT, FB-DTI and docking with AI rescoring on the high-quality manually curated testing dataset of 8 commonly used target proteins with their specific ligands.
DTI model benchmark
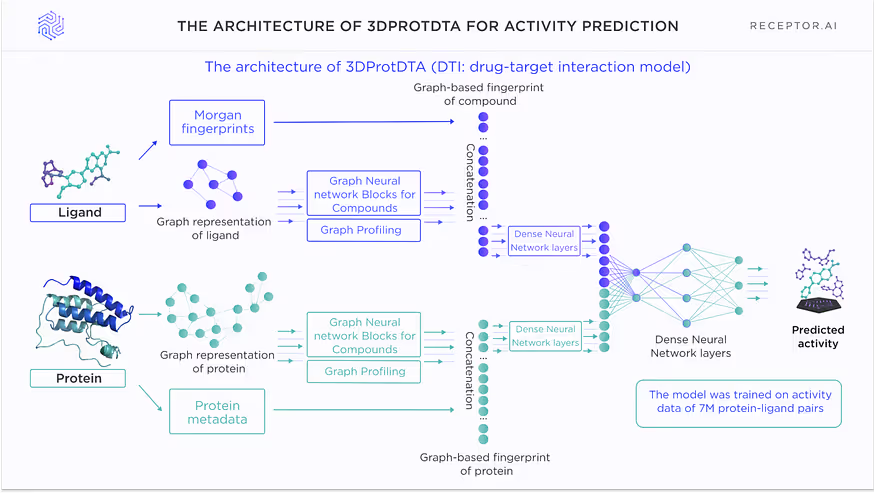
The report on the benchmarking of the DTI model (3DProtDTA, Fig.1) was given in the foregoing article, “The rock-solid base for Receptor.AI drug discovery platform: benchmarking the drug-target interaction AI model”.
FB-DTI model benchmark
Model description
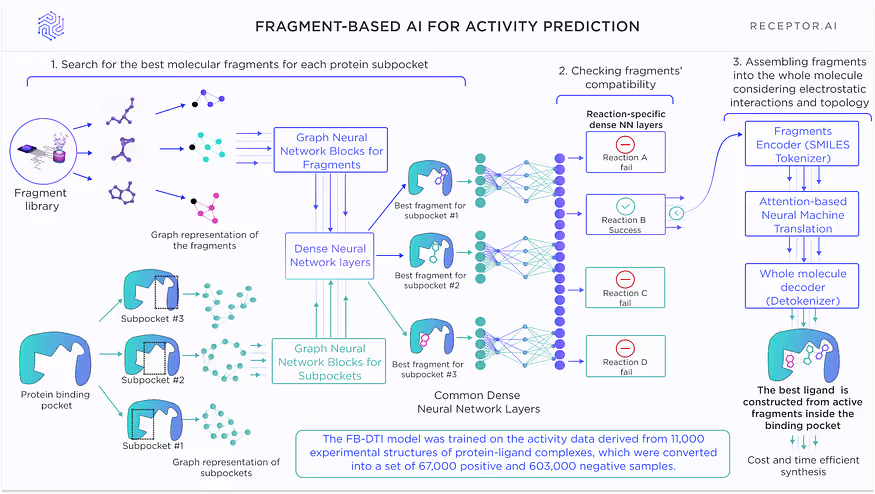
The FB-DTI model is a graph neural network model which evaluates molecular fragments against the subpockets of chosen binding pockets in the proteins of interest.
A proprietary algorithm divides the whole ligand molecule into the fragments for further evaluation by the model. Each fragment gets its own scores, which are then combined into the score of the whole molecule. This score is used for ranking candidate compounds.
FB-DTI model Architecture
The architecture of theFB-DTI model for activity prediction is shown in Fig. 2. This model requires knowledge about the concrete binding pocket but operates without any prior knowledge about existing ligands. It is used for proteins where the active site is well determined. It can operate even on proteins where no ligands are known.
Training and testing datasets
For training of the model, it used 30332 pairs of protein subpockets and hit compound fragments with experimentally validated activity and 272988 randomly generated pairs, which serve as negative controls. The test dataset contains 7583 and 68247, respectively. All the subpockets in the test dataset are retrieved from protein families, which were explicitly excluded from the training dataset (according to the Pfam protein family classification). The similarity between the testing and training datasets is 0.37(based on the Tanimoto similarity coefficient measured for 2048-bit Morgan fingerprints of radius 3).
For testing of model performance, it used an external test set of 3852 active and 11715 inactive compounds. These compounds were not used for model training.
Performance metrics
The primary performance metrics of the model are very good, but the precision-recall metrics are somewhat worse than the DTI model. This is expectable due to the specific nature of the fragment-based model, which is trained on rather small molecular fragments but used to evaluate the whole molecules by fragments (Table 1).

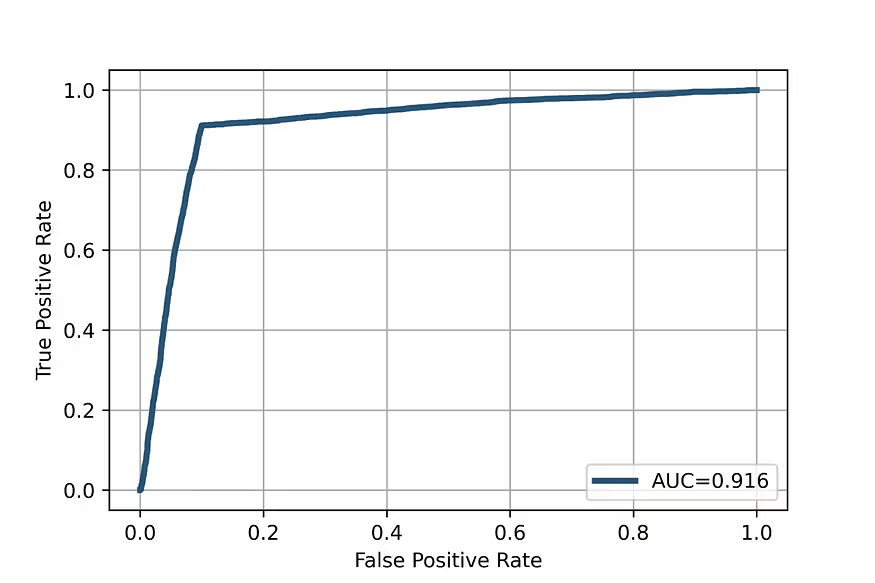
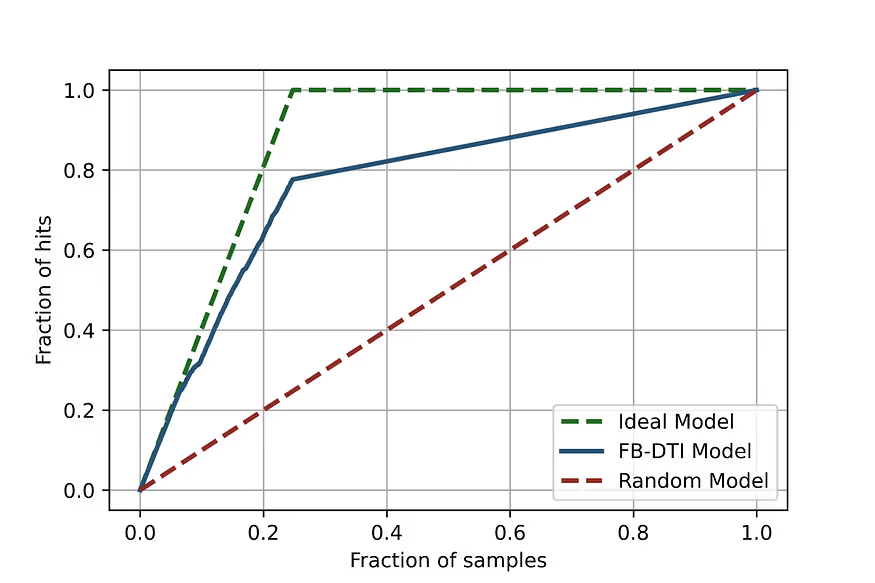
At the same time, the Receiver Operator Characteristic curve, which is the most important performance characteristic of this kind of model, is very good (Fig. 3) and the enrichment plot shows that the model prioritises the hits successfully (Fig. 4).
Docking rescoring model
Model description
The docking rescoring AI model operates on the docking poses obtained by a traditional AutoDock-based docking engine. The AutoDock scores are not used, and the top poses of the docked ligands are rescored and reranked by this model. This model is used in the final stage of secondary virtual screening in order to rank the best candidate compounds.
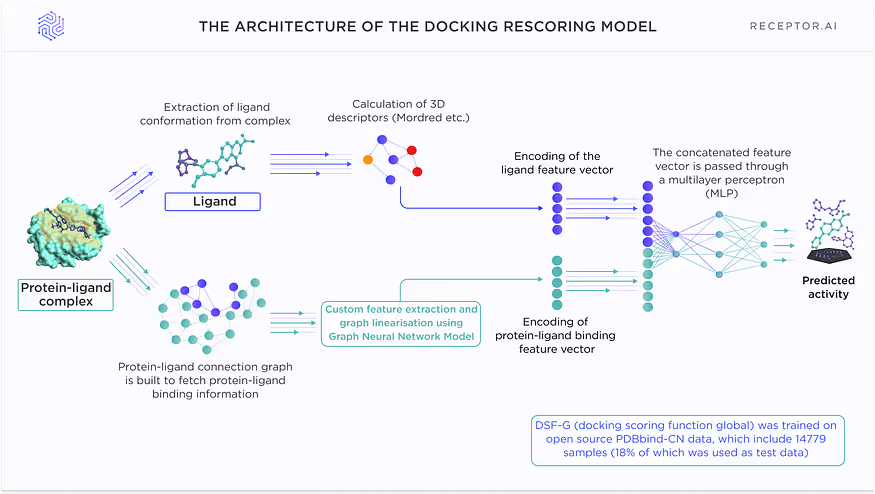
The model is based on a proprietary graph neural network that encodes protein-ligand connectivity graphs, and the Mordred descriptors for the ligands are used. The protein-ligand connectivity graph is passed through the GNNs layers, then encoded as a 320-dimensional vector with a 456-dimensional feature vector of Mordred descriptors and passed through the multilayer perceptrons (MLPs) to get the final predictions.
Docking rescoring model
The architecture of thedocking rescoring model is shown in Fig. 5. This model takes the docking poses obtained by conventional docking and rescores them in accordance with predicted activities. The model is trained on all available protein-ligand complexes for which the activity data are known. The model encodes connectivity graphs of the protein-ligand complexes and 3D conformation-dependent descriptors of the ligands themselves.
Training and testing datasets
The model is trained on the ligand activity data taken from the open-source PDBbind-CN database. The training dataset contains 14779 protein-ligands complexes, while the testing dataset contains 3239 protein-ligand pairs (~18% of all data), which were explicitly excluded from the training data. The similarity between the training and testing datasets was 0.45 (computed as the Tanimoto similarity coefficient based on a comparison of Morgan 2048-bit fingerprints with radius 3).
Performance metrics
The primary performance metrics of the model are shown in Table 2 and Figures 6 and 7. We made a comparison of the manually tuned AutoDock scoring functions, which are used internally in our docking engine, and the AI-based docking rescoring function on the same dataset of the ligand-protein complexes. The actual poses of the ligands were taken from the dataset itself, and the scoring functions were used to rank them.
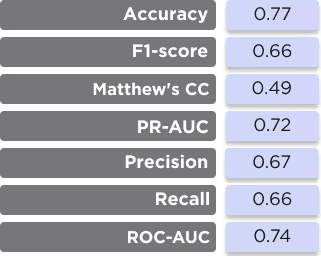
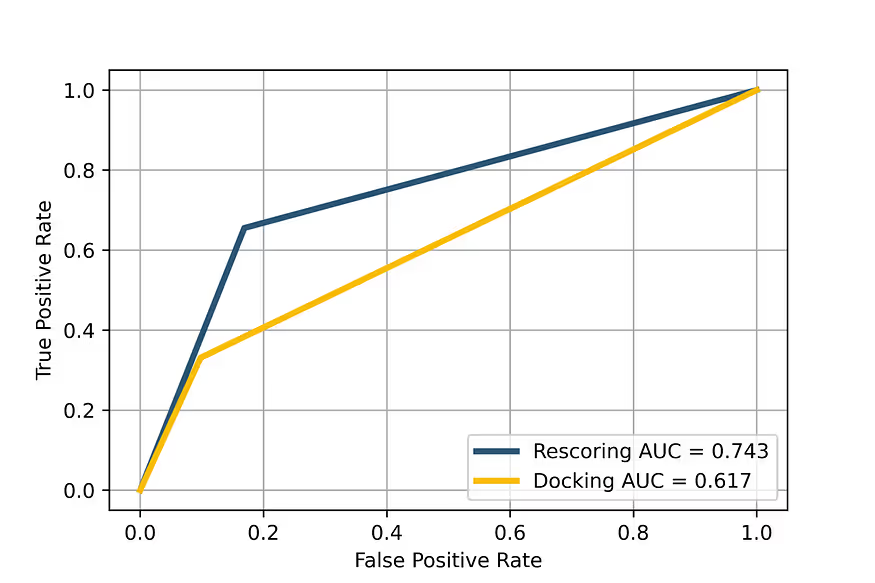
Despite the relatively small number of available samples, the model gives good performance metrics for predicting the real activity of the docking poses. Although the absolute values are not ideal, they are enough for practical application. It is necessary to note that this model is used for ranking predicted binders, so the exact relative position of the compound is not critically important if a sufficient number of compounds is used for subsequent biological testing. At the same time, the model correctly ranks the compounds which are different in activity by order of magnitude or more, which is critically important for providing correct information to the general consensus function.
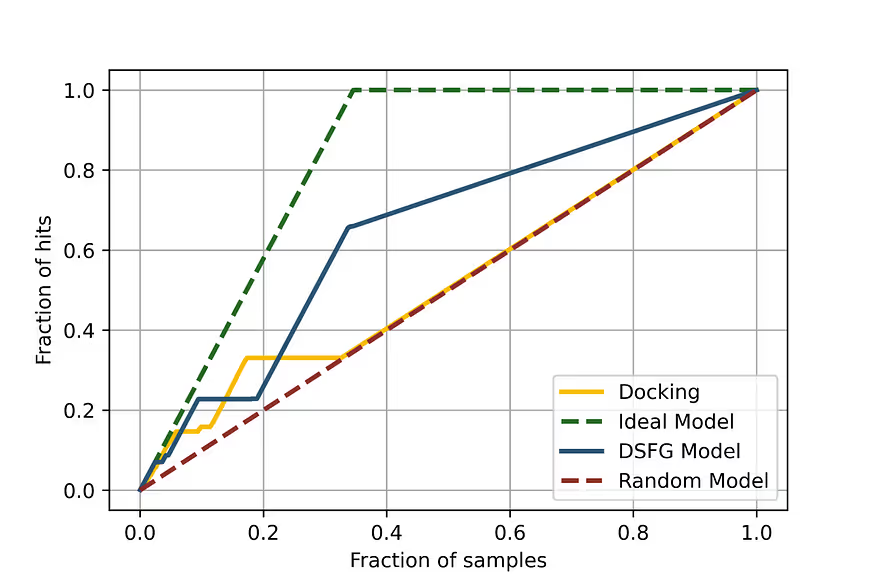
The results suggest that our docking rescoring model performs very well in ranking the docking poses according to real poses of compounds in the protein-ligand complexes with the structure.
Benchmarking the consensus function and comparing with docking techniques
Benchmark description
A molecular docking is routinely used in the later stages of virtual screening in order to prioritise and rank the promising compounds according to their binding affinity, which is estimated by means of the docking score based on the sum of interatomic interactions (the docking force field). Although such a technique is physics-based and thus applicable to a wide variety of target proteins and ligands, it has several limitations, such as limited performance and the inability to assess the biological activity beyond the binding strength. Despite these drawbacks, the docking is still considered a “golden standard” technique by many drug discovery professionals.
In order to demonstrate how our AI-based methods compare with existing docking techniques, we used a set of 4 well-known protein targets: Carbonic anhydrase II (CA2), Androgen receptor (AR), Cathepsin D (Cath-D), Beta-secretase 1 (BACE1) and Janus kinase 1 (JAK1). We created a manually curated dataset of known active ligands for these proteins for which the binding affinities are determined with high confidence and low probability of error (see Table 3 for the number of selected ligands).
We applied five different techniques: DTI, FB-DTI, docking with AI rescoring and a smart consensus function based on several techniques (combination of DTI, FB-DTI and docking with AI rescoring), in order to rank the compounds for each protein according to predicted binding propensity. We also extracted literature data for the same ligands for 16 widely used docking techniques and compared them with the results of our methods.
Limitations
There are several limitations in such comparison, which should be emphasised before presenting the results:
- DTI and FB-DTI models are designed for the initial stage of virtual screening. They are optimised for discriminating binders from non-binders and not for the accurate ranking of the proven binders. That is why comparing them to docking techniques, which are used in the secondary screening stage, is not completely fair.
- All our AI models are trained on activity data. Although the binding affinity correlates with biological activity for the majority of active compounds, our models are not tuned to predict affinity itself.
Due to these limitations, the results should be considered semi-quantitative only. Nevertheless, they provide valuable information about the performance of our techniques on ranking the known binders for several classical drug discovery targets.
Performance metrics
Figure 8 shows the average r2 scores of all compared techniques.
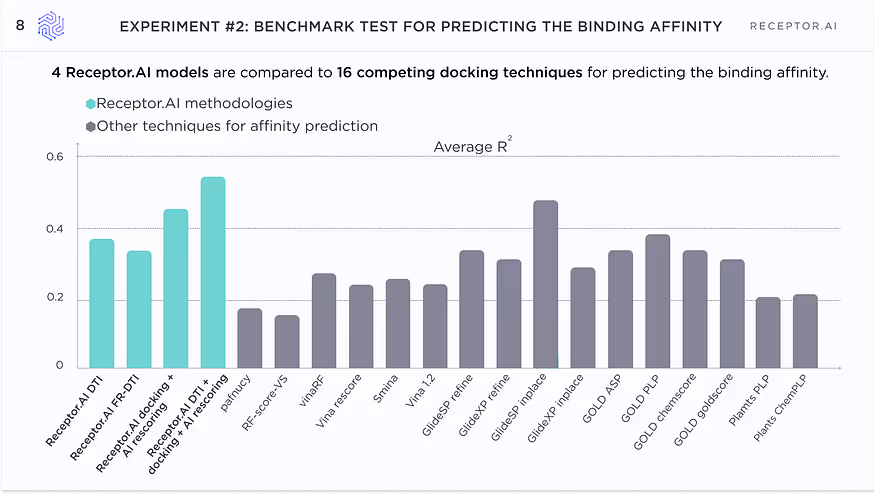
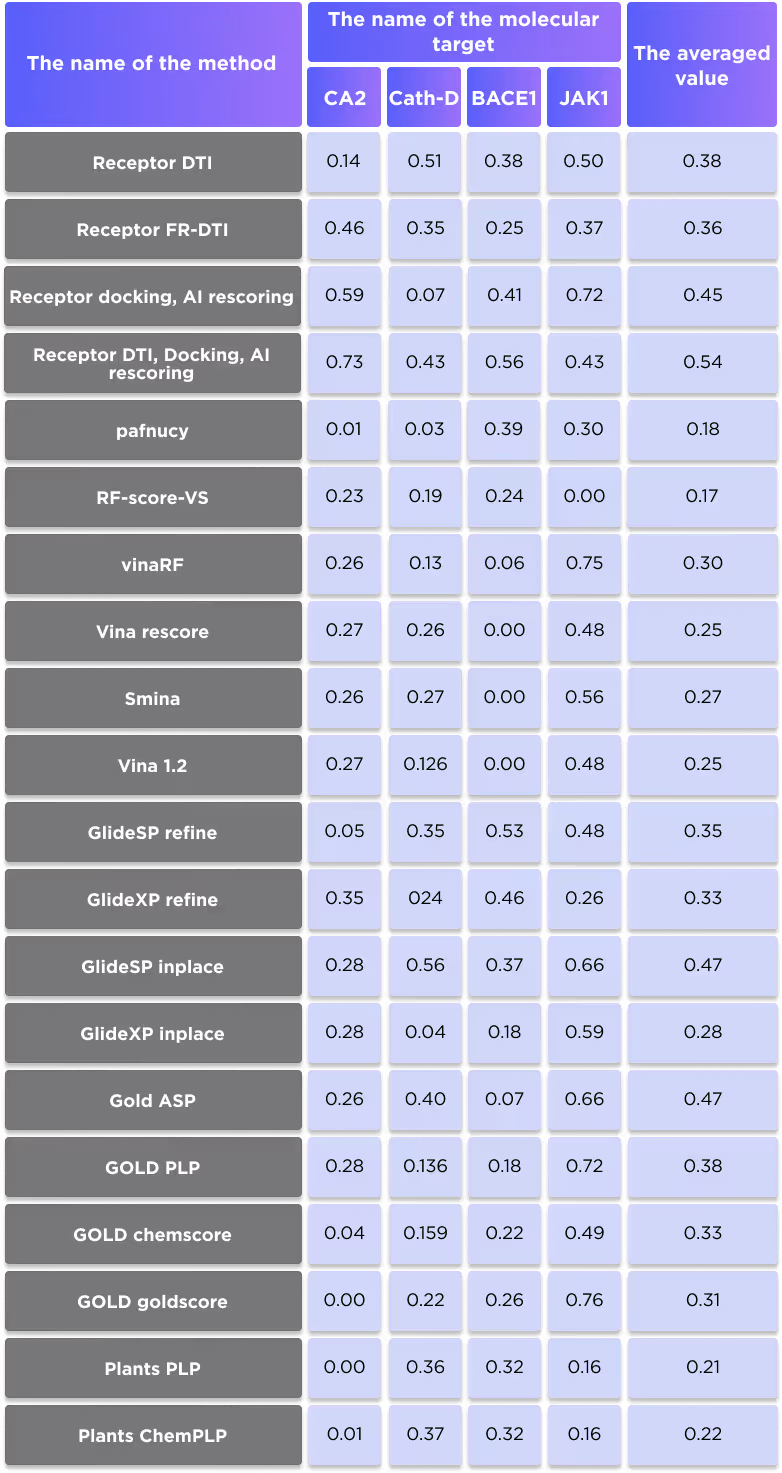
It is clearly seen that even the DTI and FB-DTI techniques, which are not intended for ranking of the known binders, perform on par with dedicated docking techniques and only slightly worse than the most accurate docking methods. Our docking with AI rescoring shows even better results, which are on par with the best docking techniques, such as GOLD and GlideSP. Finally, the combination of DTI and docking with AI rescoring outperforms all studied docking techniques.
This gives us confidence that our methods not only correctly discriminate binders from non-binders but also rank the binders with high accuracy, which is comparable to or even better than one provided by dedicated docking techniques.
Conclusions
The structure-based techniques developed by Receptor.AI for the secondary stage of virtual screening demonstrate the solid performance of activity prediction. They also perform on par with the best-dedicated docking techniques for determining binding affinity. However, the best results are achieved with a smart consensus function, which combines the scores obtained by all AI techniques (DTI, FB-DTI and docking with AI rescoring) into a single quantitative scale of predicted activities. Choosing the functional form of a consensus function and its parameters adds another layer of optimisation, which, however, leads to the most accurate and robust results.
That is why these techniques form a reliable basis for target-specific screening in the Receptor.AI drug discovery platform.