Designing protein proximity inducers with Receptor.AI drug discovery platform
Announcement


Full Text
Protein proximity inducers with different mechanisms of action are emerging as promising alternatives to conventional small-molecule drugs with many potential advantages. Despite their growing popularity, very few tools on the market address the rational design of such compounds with dedicated workflows that take into account all their specific features.
Receptor.AI is the first company that developed an AI-based drug discovery platform specifically tailored for designing proximity-inducing small molecules and peptides. We design the compounds that alter the protein-protein interactions in multiple ways: molecular glues, different types of degraders, protein localization modulators, and more. We use the power of our highly performant and well-tested workflow for small-molecule drug discovery to design monofunctional glue compounds and individual structural parts of bifunctional compounds with different mechanisms of action.
1. Protein proximity inducers types
There are two types of proximity inducers:
1. Bifunctional inducers
a. Proximity engagers
b. Degraders
c. Protein localization modulators
d. FKBP sequestrators
2. Mono-functional inducers
a. Molecular glues
b. PPI stabilizers
c. PPI disruptors (reversed inducer)
1.1 Bifunctional protein degraders
Proteolysis-targeting chimeras (PROTACs) and lysosome-targeting chimeras (LYTACs) leverage intracellular degradation pathways to achieve targeted protein degradation, presenting a therapeutic strategy superior to conventional inhibition in multiple aspects. For example, unlike traditional inhibitors that bind to a single copy protein globule, thus requiring high doses with potential off-target effects, PROTACs and LYTACs induce the degradation of multiple copies of the target protein. This mode of action not only minimizes the required dosage but also reduces the likelihood of drug resistance and toxicity.
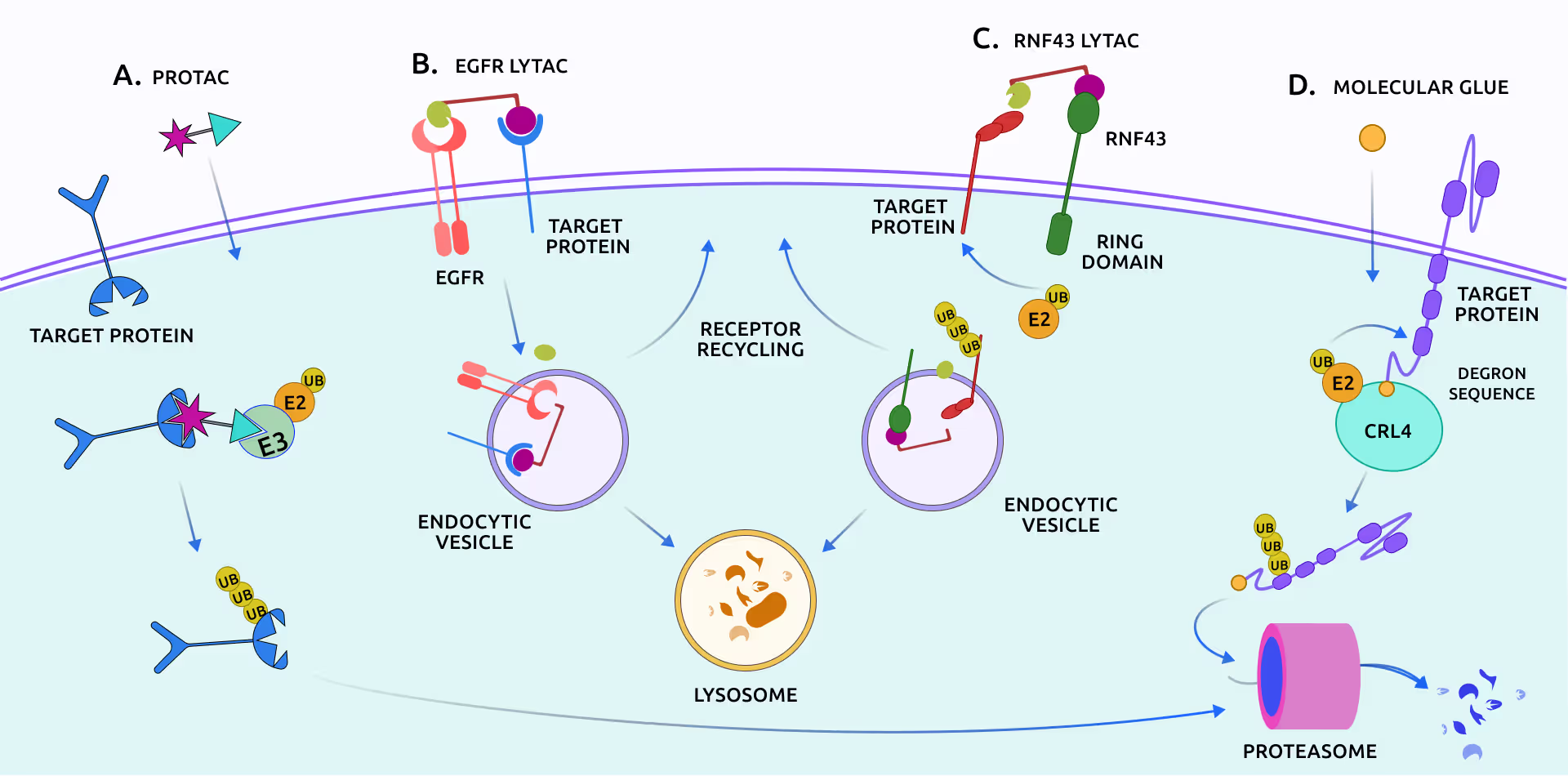
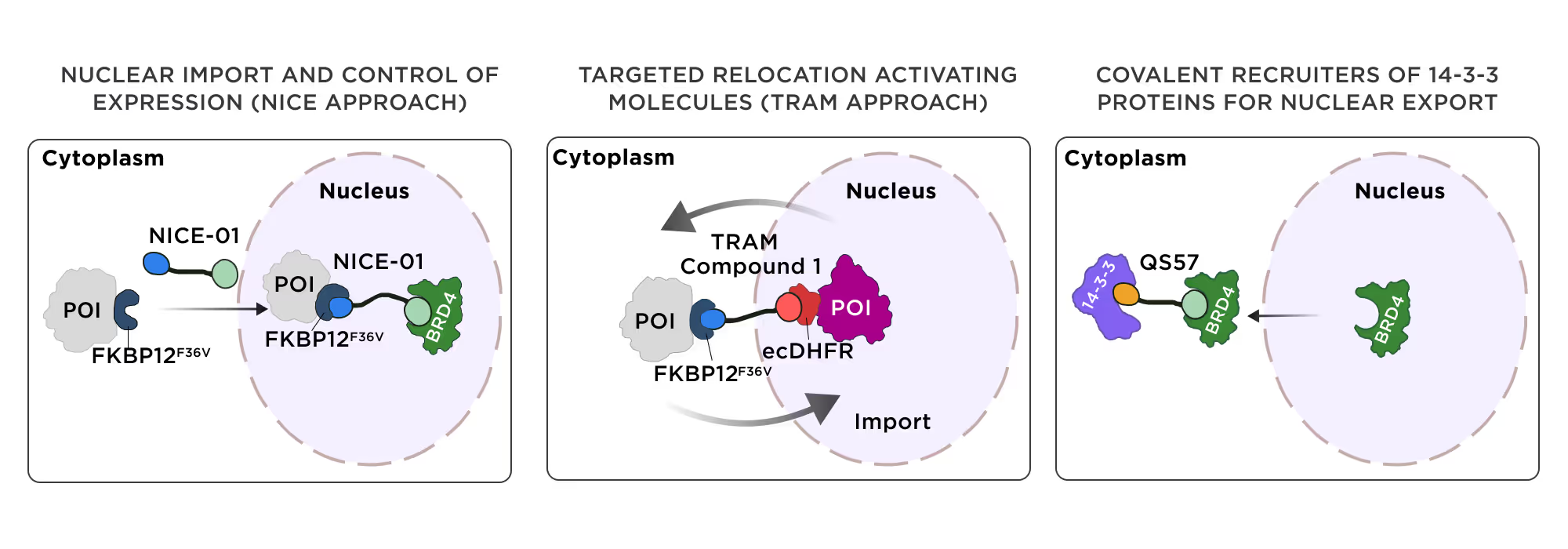
1.2 Bifunctional compounds for protein relocalization
Targeted relocalization of therapeutically important proteins is an emerging trend in drug discovery, which exploits the cellular protein compartmentalization and trafficking mechanisms. The protein of interest could be forcefully transported out of its native cellular compartment, thus suppressing its function. Alternatively, the natural clearance of the target protein from its functional compartment could be suppressed to enhance its action. Exploiting these mechanisms requires attaching the protein of interest to the dedicated transporters or signaling molecules using bifunctional compounds in covalent or non-covalent ways.
1.3 Bifunctional compounds mimicking antibodies
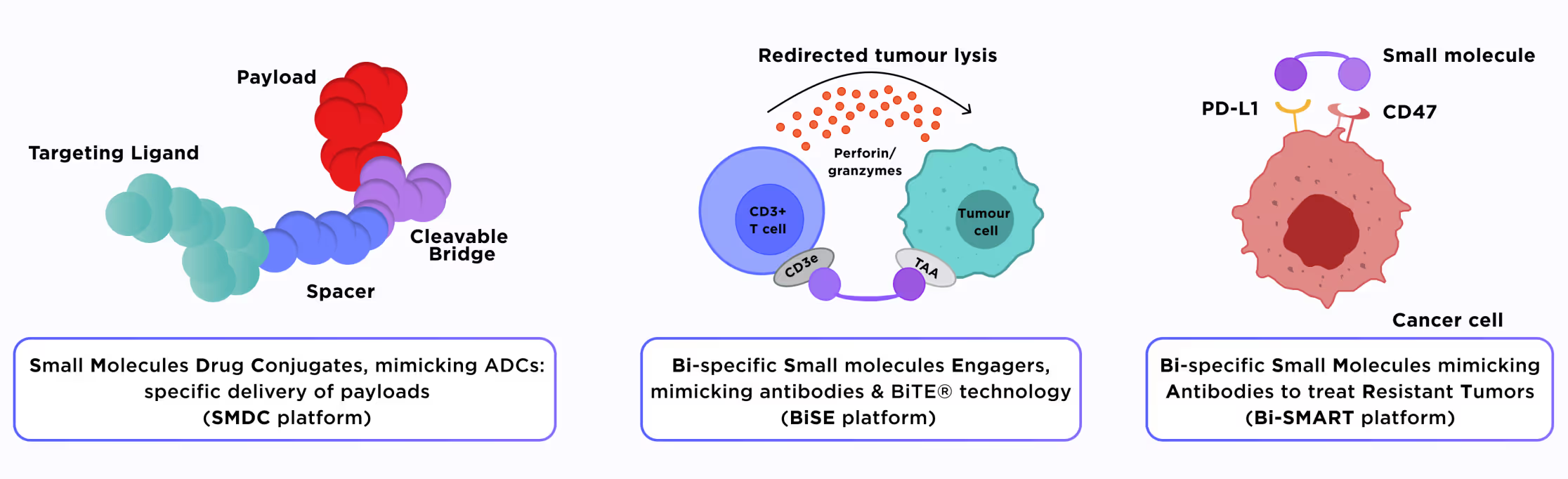
Antibodies are great therapeutic tools due to their ability to link together their specific epitopes. However, they possess several drawbacks, such as lack of oral administration, poor permeability into solid tissues and tumors, immunogenicity, etc. Bifunctional small molecules could mimic the cross-linking abilities of antibodies without their specific drawbacks. Having two specific warheads, they can link together two proteins located on the same or different cells. It is also possible to design the small molecule drug conjugates (SMDCs) in the same manner as antibody-drug conjugates (ADCs) are made. In this approach, one part of the conjugate acts as a selective anchor, while the other part is a payload, which has to be delivered to its target.
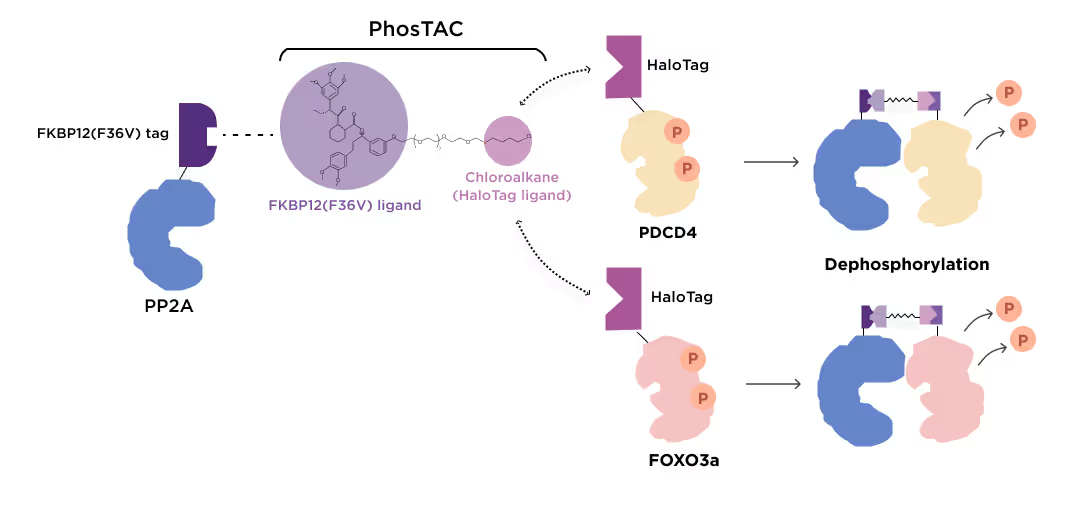
1.4 Modulation of Protein Activity by Phosphorylation Targeting Chimeras (PhosTACs)
PhosTACs are a promising emerging modality of manipulating protein activity by recruiting kinases and phosphatases into the physical vicinity of the protein of interest using bifunctional proximity engagers. The target-agnostic nature of the Receptor.AI platform allows for discovering potential PhosTACs by designing specific warheads binding to the protein of interest and required kinase of phosphatase.
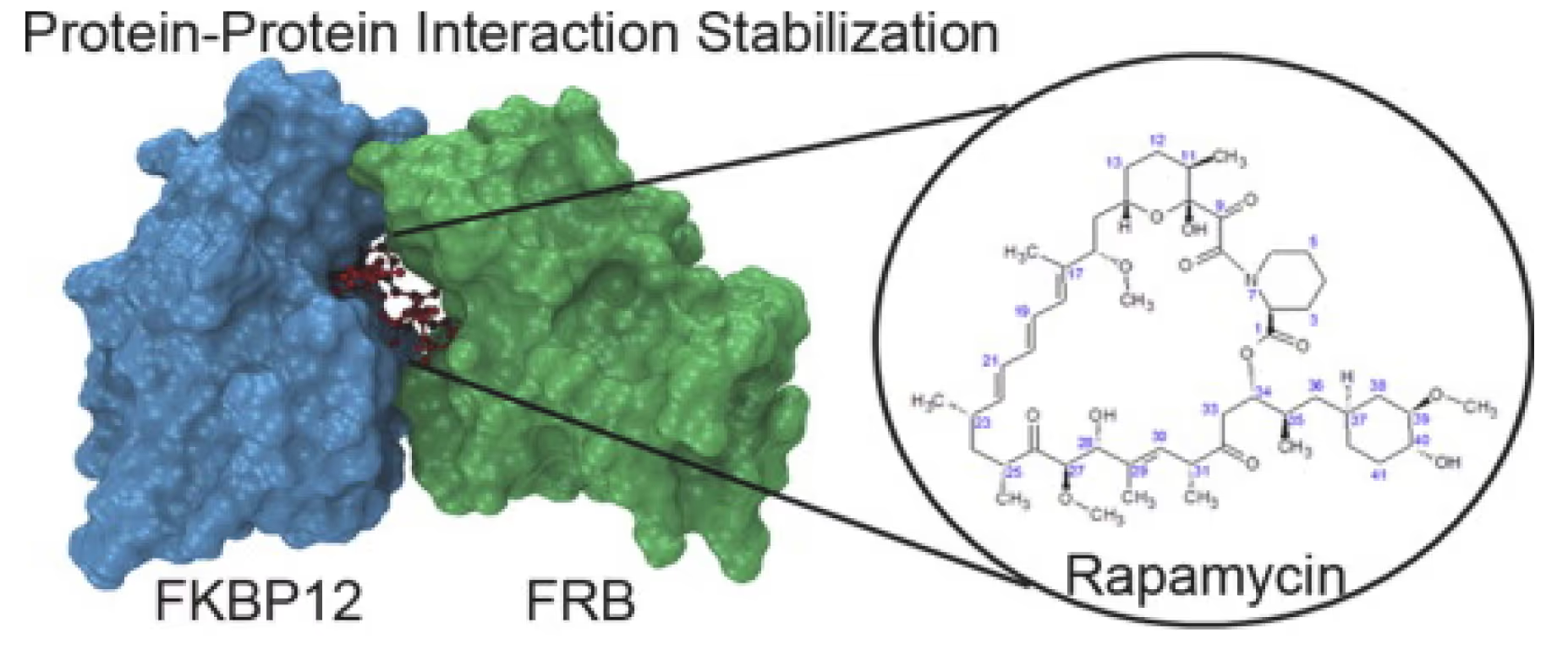
1.5 FKBP-based protein sequestration
Gluing proteins with FKBP utilizing its highly specific binding to rapamycin-like molecules is a promising new strategy for sequestering unwanted proteins and thus reducing their activity in cells. Rapamycin itself selectively glues FKBP to mTOR only. Our platform is capable of designing rapamycin functional derivatives that are able to glue FKBP to other proteins as well allowing for a viable alternative to protein degraders.
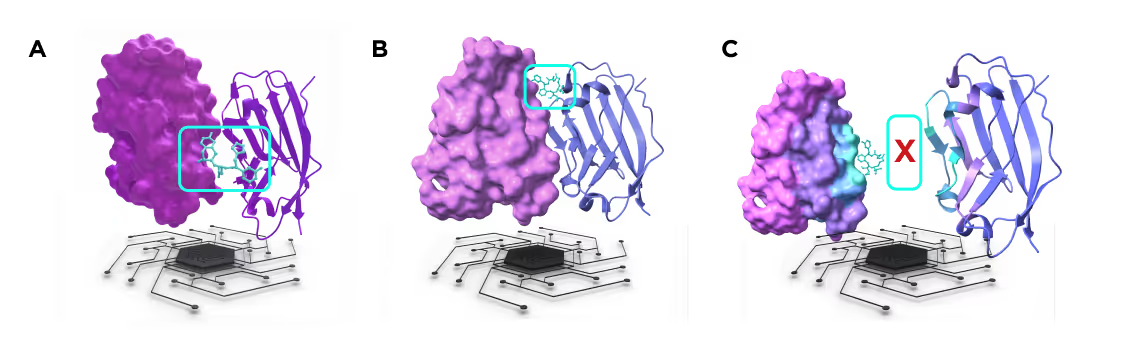
1.6 Monospecific proximity inducers
Molecular glues
Molecular glues are compounds that bring together two proteins that are otherwise not forming a complex at all or only interact transiently. The molecular glues may have different modes of action but their design follows the same principle - the molecule should bind strongly to both partner proteins while having a compact molecular structure. This is in contrast to bifunctional compounds that typically possess two distinct warheads separated by an extended linker.
PPI stabilizers
PPI stabilizers are compounds that make existing physiological protein-protein interactions stronger and shift the equilibrium between bound and dissociated states toward the complexes. Stabilizers could be either topologically compact (similar to molecular glues) or extended with two distinct warheads binding to the partner proteins at a significant distance (bridging molecules).
PPI disruptors
PPI disruptors are opposite to PPI stabilizers. They make existing physiological protein-protein interactions weaker and lead to dissociation of the protein-protein complexes. Disruptors may have two distinct modes of action: (1) binding to individual partner proteins before complex formation and blocking the interaction interface or (2) binding to an already formed complex and destabilizing it by inducing allosteric conformational changes or competing with the residues forming the protein-protein contacts.
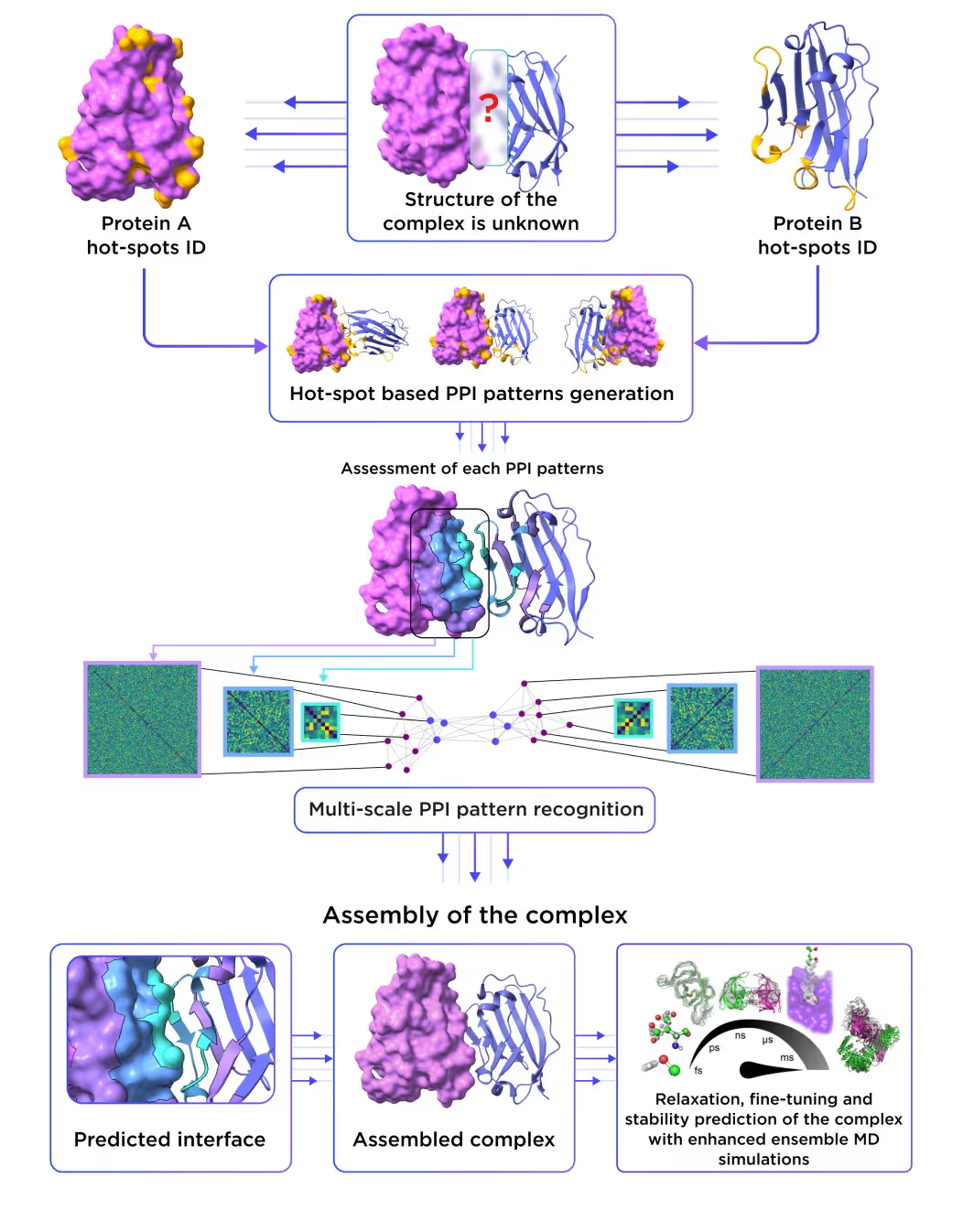
2. Receptor.AI’s template-agnostic PPI model excels in predicting protein-protein and protein-nucleotide complexes, surpassing AlphaFold3.
The figure below (Fig. 7) showcases the architecture of our template-agnostic PPI prediction model. Leveraging cutting-edge data augmentation algorithms, this model was trained using the largest PPI dataset providing precise insights into protein interactions and revolutionizing the design of proximity inducers. The model surpasses AlphaFold3 in terms of accuracy and works even in the case when no structures of homologous complexes are resolved experimentally.
3. Receptor.AI platform for designing bifunctional compounds
Receptor.AI platform allows designing of bifunctional compounds using a dedicated workflow. The platform utilizes the paradigm of an iterative design-test-modify development loop, where experimental feedback is incorporated into the design workflow on each iteration.
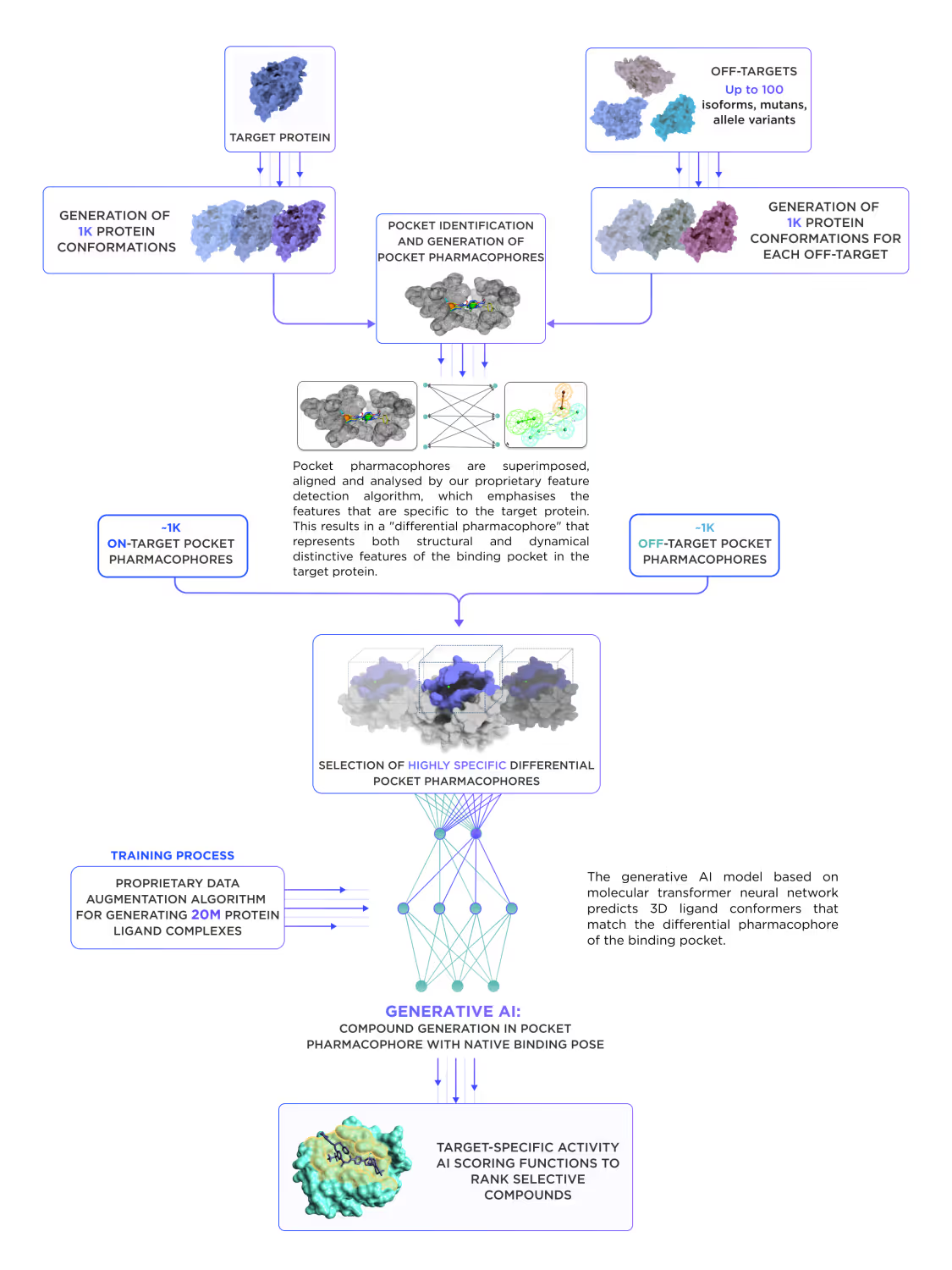
3.1. Warhead/anchor design
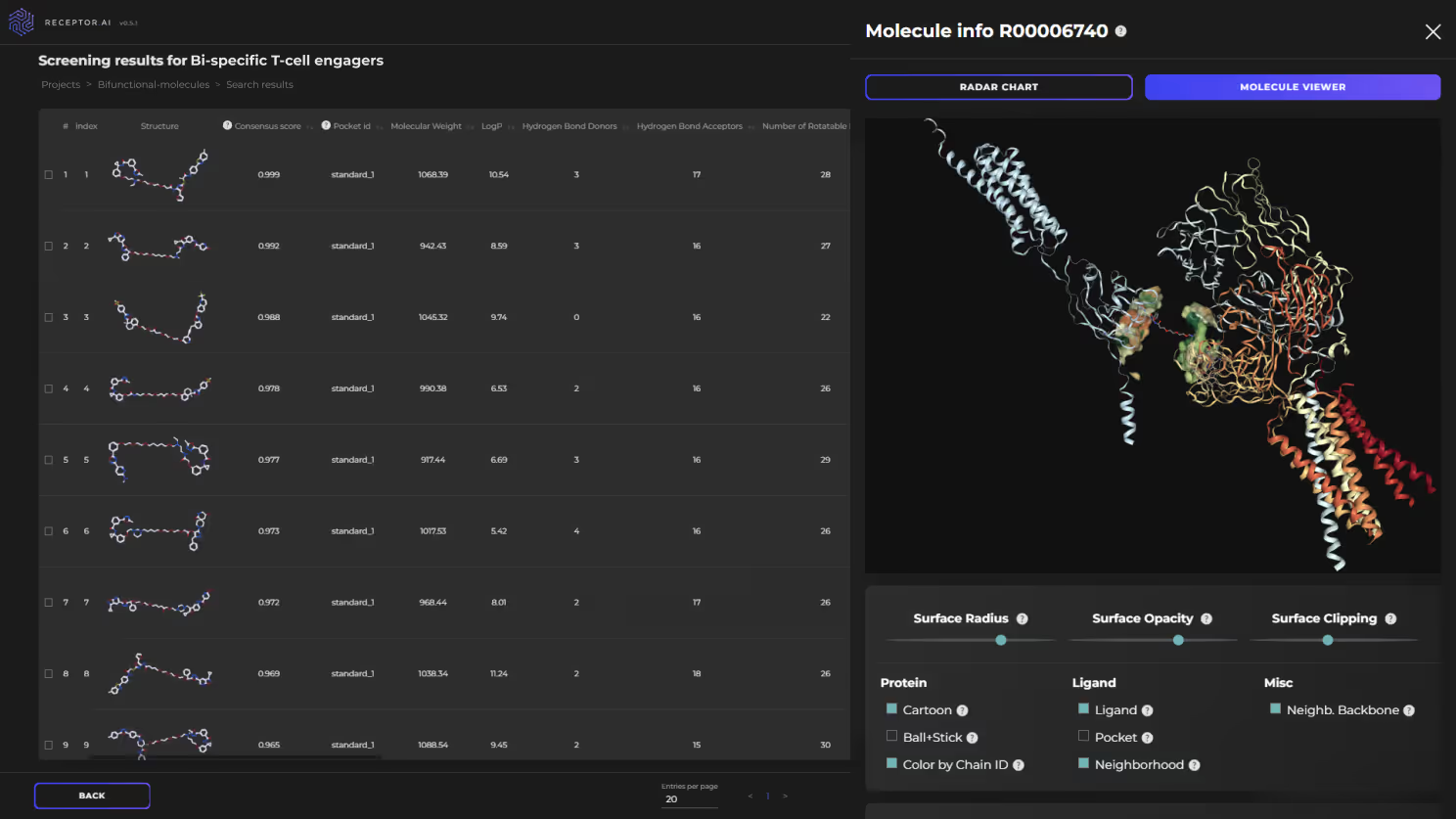
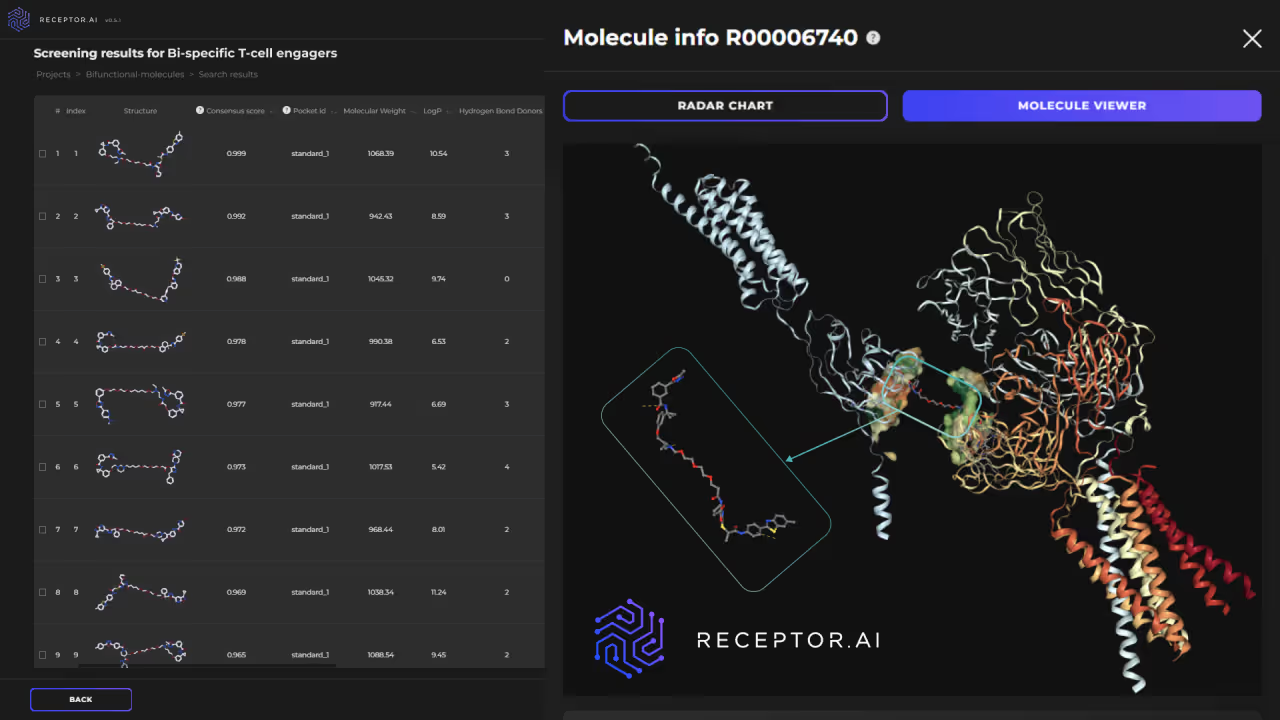
We start with predicting the most promising binding pocket for the warhead binding on the protein of interest (POI). This may or may not coincide with the functionally active sites and is facilitated by a dedicated AI model. In parallel, the site for anchor binding is identified on the desired partner protein. Both POI and the partner protein are subject to initial virtual screening using the dedicated module of our platform (Fig. 8), which incorporates affinity prediction AI models and a proteome-wide selectivity prediction. Afterward, selected candidate compounds are forwarded to the secondary screening models, which filter them by over 60 ADME-Tox endpoints, physicochemical parameters, and drug-likeness metrics. The docking with AI scoring follows, prioritizing the best candidate compounds.
At this stage, we can also incorporate available data from high-throughput experiments such as DEL screenings. A big advantage of the Receptor.AI approach to PROTACs and LYTACs design is the usage of a well-tested AI platform, which was validated on multiple small molecule drug design projects. The warhead and anchor moieties of bifunctional compounds are typical small drug-like molecules combined by a linker. Thus, the small molecule design workflow could be used separately for them in the first stage. After that, the experimental validation of warhead affinity to POI and the anchor affinity to the ligase is performed, and the results are used to retrain and fine-tune AI models for subsequent iterations.
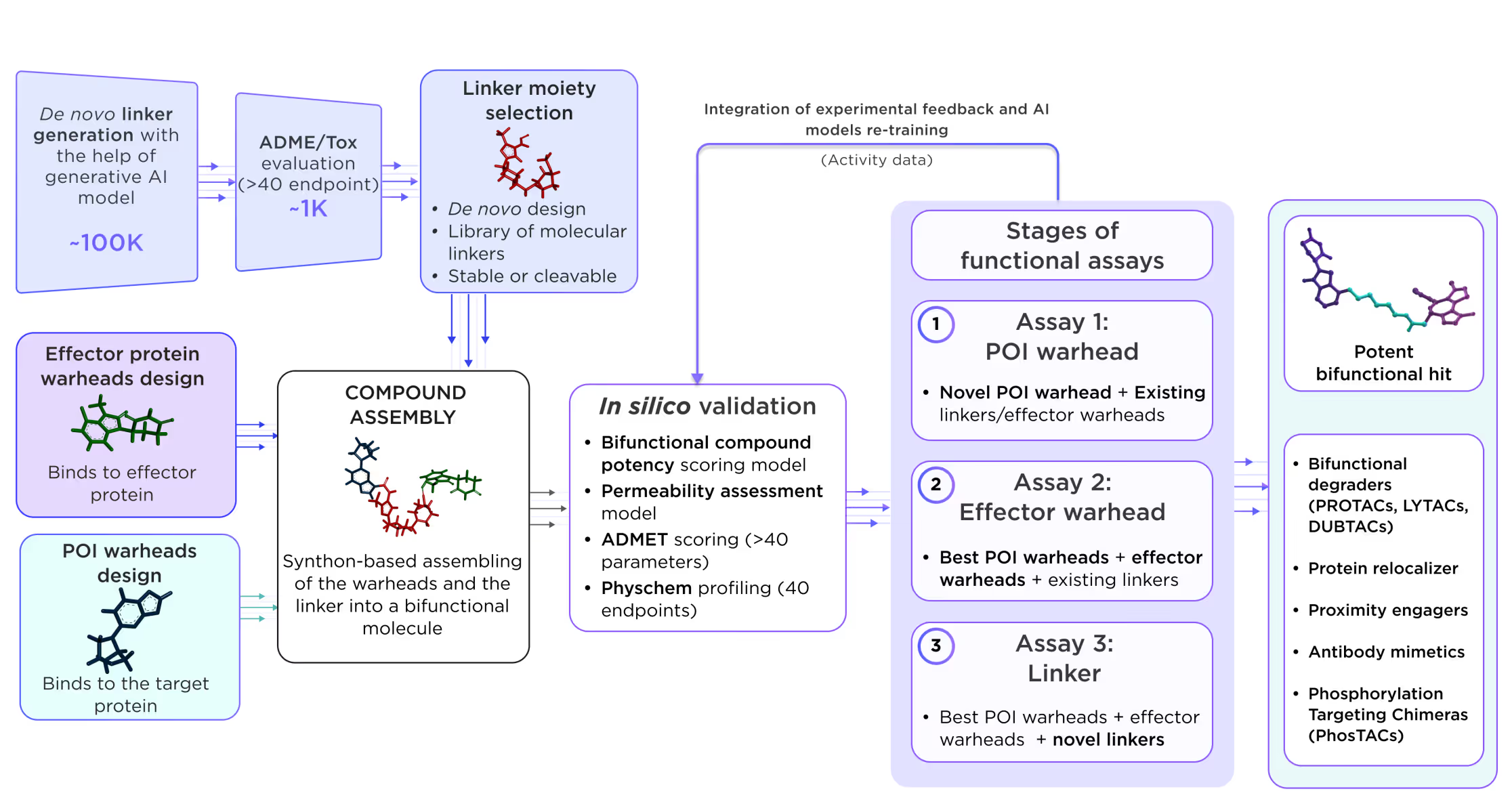
3.2. Linker design
In parallel, the linker design path is followed. The system can either utilize known linkers or design the linker de novo using generative graph neural network models with rigidity and length constraints. Prospective linkers are assessed with the ADME-Tox and drug-likeness filters to ensure their safety. The custom design of the linkers can also be performed a posteriori in the later stages after collecting experimental data from assembled bifunctional compounds. The classification AI model can be trained on the most successful anchor-linker-warhead combinations. Data on existing linkers with custom-tuned length and rigidity can also be used. As a result, the model will determine the most promising linkers.
3.3. Assembly and whole-molecule optimization
The best warheads, anchors, and linkers are then subject to combinatorial in silico synthon-based assembly into whole bifunctional molecules. Up to 10 of the most promising novel, assembled compounds could be further assessed by MD and FEP methods. Selected candidate molecules are transferred for experimental validation using functional assays, and their results are fed back to the platform for AI model retraining and tuning (if needed). We propose an iterative validation process, which increases the success rate by decreasing the number of tested variables on each iteration (Fig. 9).
3.4. Membrane permeability prediction
In addition to this standard procedure, which roughly follows the workflow for small molecules, special attention has to be paid to the membrane permeability of designed bifunctional molecules. Being large and bulky molecules, they do not easily cross the membrane unless designed to do so from the ground up. We are leveraging the proprietary technology of designing membrane-targeting drugs, which is being developed in Receptor.AI, to ensure that the designed chimeras are amphiphilic molecules with a high propensity for membrane crossing.
Conclusions
Recently proximity inducers emerged as a novel and promising drug modality with promising applications within the pharmaceutical and biotechnology industries. However, the absence of defined methodologies for their development slows their adoption and usage.
In Receptor.AI we integrated our forefront models into the platform such as:
- Template-agnostic PPI prediction.
- Advanced selectivity enhancement.
- Bifunctional compound assembly and permeability prediction.
The whole integrated system, powered by a proprietary LLM engine, automates the proximity inducer design process.