
LLM-driven binding pocket prioritization
Announcement


Full Text
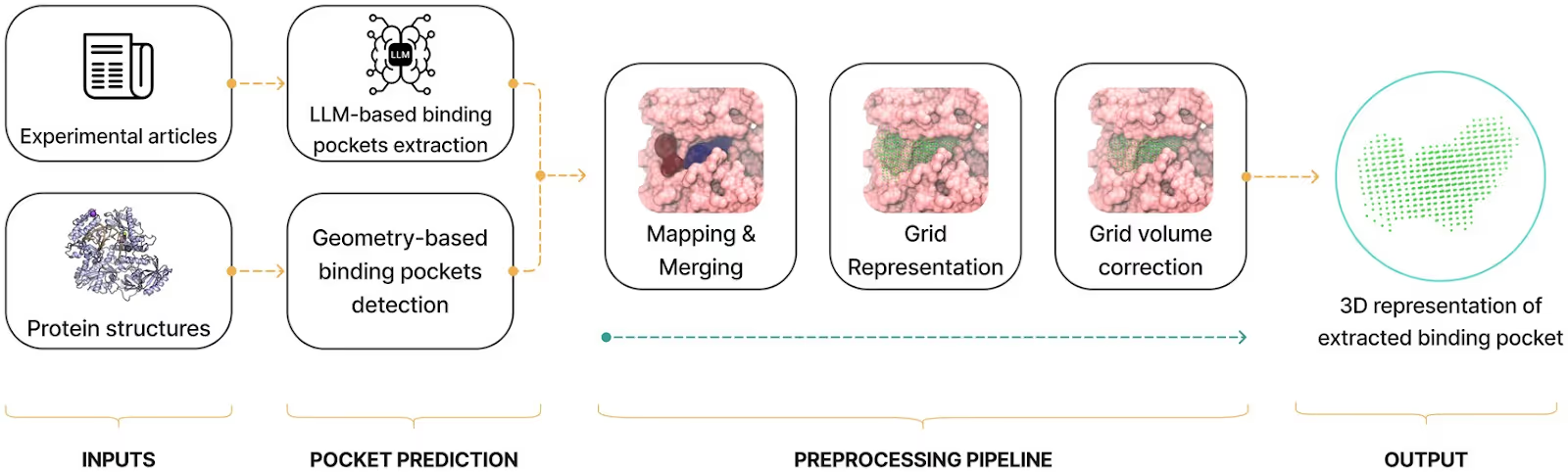
Binding pocket identification is a cornerstone of structure-based drug discovery. Geometry-based tools such as Fpocket efficiently detect cavities on protein surfaces, but they tend to overpredict, producing dozens of candidate sites with no guarantee of biological relevance. Traditionally, researchers filter these predictions by reading papers and compiling residue-level binding annotations — a process that is slow, inconsistent, and difficult to scale.
Large language models (LLMs) now offer a way to automate this step. By extracting and structuring binding-site evidence from the literature, LLMs can refine geometry-based predictions into biologically validated pocket models.
Benchmarking the Workflow
We developed and tested a hybrid pipeline that combines Fpocket cavity detection with LLM-driven literature mining. Benchmarks were run on ten diverse proteins and 31 annotated publications.
Geometry only (Fpocket): many candidate sites, but frequent false positives.
Hybrid LLM workflow: Pocket Number Accuracy improved to 0.71 vs. ~0.5 baseline, recall remained perfect at 1.0 (no true sites missed), and specificity increased from ~0.46 to ~0.66.
The result: every biologically validated pocket was captured, while spurious cavities were systematically filtered out.
From Text to 3D Geometry
The core innovation is mapping residue lists mined from publications onto 3D structures. Overlapping sites are merged, fragmented pockets recombined, and cavities trimmed by convex hulls around literature-derived residues.
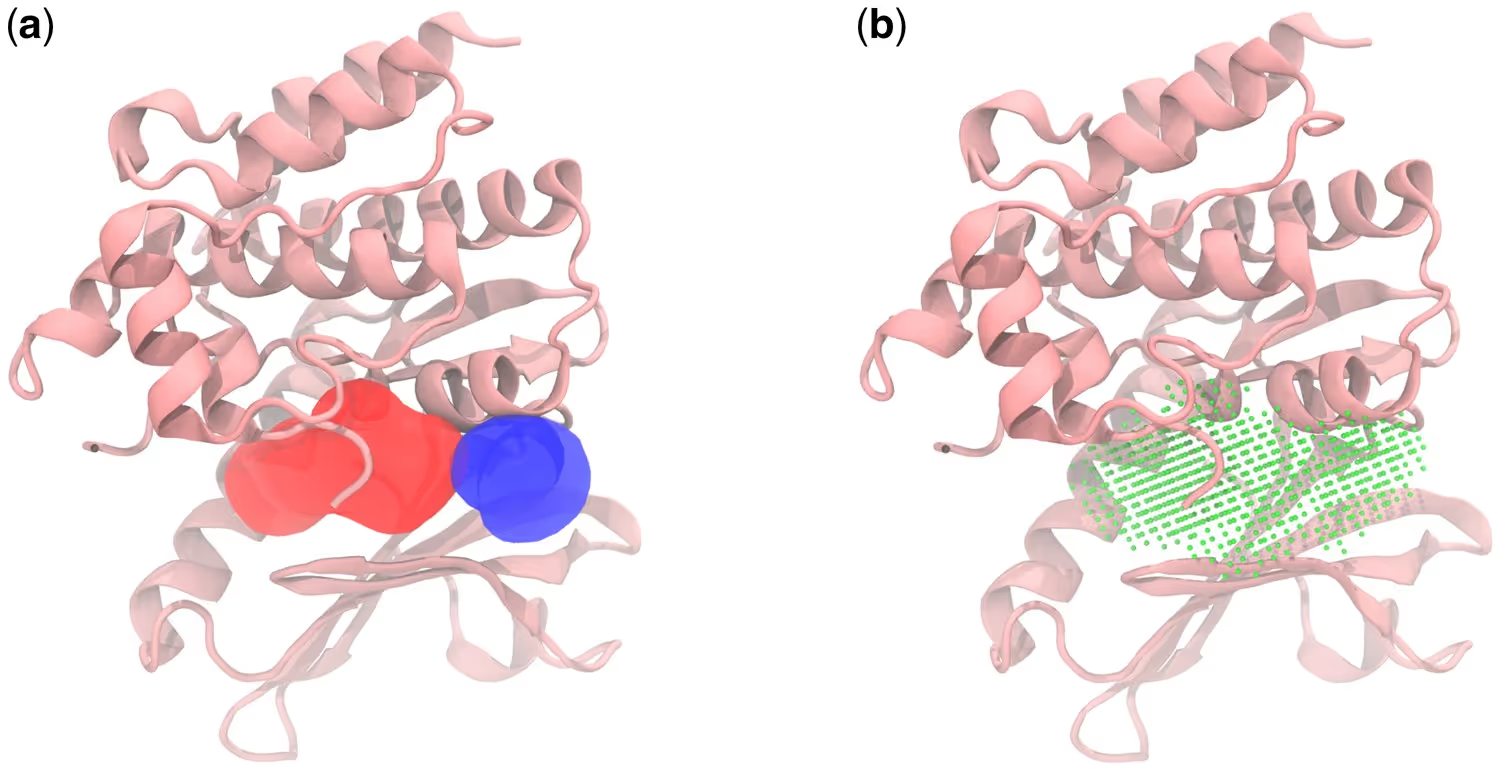
In multimeric proteins, the method also recovers interface sites. For example, the GABAA receptor contains two symmetric anesthetic-binding pockets at different subunit interfaces. Fpocket detects both but cannot establish equivalence. Literature-mined evidence confirmed that etomidate binds in both, ensuring they were correctly annotated as validated sites.
Conclusion
LLMs add a missing layer to structure-based drug discovery — a direct link between decades of experimental evidence in the literature and computational predictions. By combining geometric algorithms with literature mining, pocket models become not only geometrically plausible but also biologically validated.
This hybrid approach transforms literature into data, making drug discovery workflows faster, more reliable, and more scalable.
Resources & Links
Our article: Leveraging large language models for literature-driven prioritization of protein binding pockets. https://doi.org/10.1093/bioinformatics/btaf449