
PPI modality: How can AI overcome data scarcity?
Announcement


Full Text
Protein–protein interactions (PPIs) are essential in biology and drug design. Knowing the structure of a protein–protein complex, we can design stabilizers, inhibitors, or molecular glues.
Receptor.AI’s CEO recently presented at the Promega TPD Symposium, where he outlined current challenges with PPI modalities in drug discovery. The main topics were:
- PPI experimental data scarcity;
- co-folding and docking limitations;
- proposed solutions.
The PPI problem — Scarcity of Experimental 3D Data
BioGRID database curates evidence records for over 2.2 million PPIs, yet only around 23,000 complexes have resolved 3D structures. Moreover, known structures are biased toward stable, soluble, globular assemblies, whereas most PPIs are transient, involve intrinsically disordered regions, or occur at membranes.

Resolving protein–protein complexes is challenging. Experimental methods such as X-ray crystallography or cryo-EM are expensive, slow, and sometimes impossible for flexible or membrane proteins. That’s why computational prediction is critical to fill this gap.
PPI 3D structure prediction is currently presented by two approaches: co-folding and docking. Protein–protein docking has evolved over the past 20 years and remains more accurate. Protein–protein co-folding has emerged recently and is improving faster. But can they meaningfully compete with experimental methods?
Co-folding and Docking Limitations
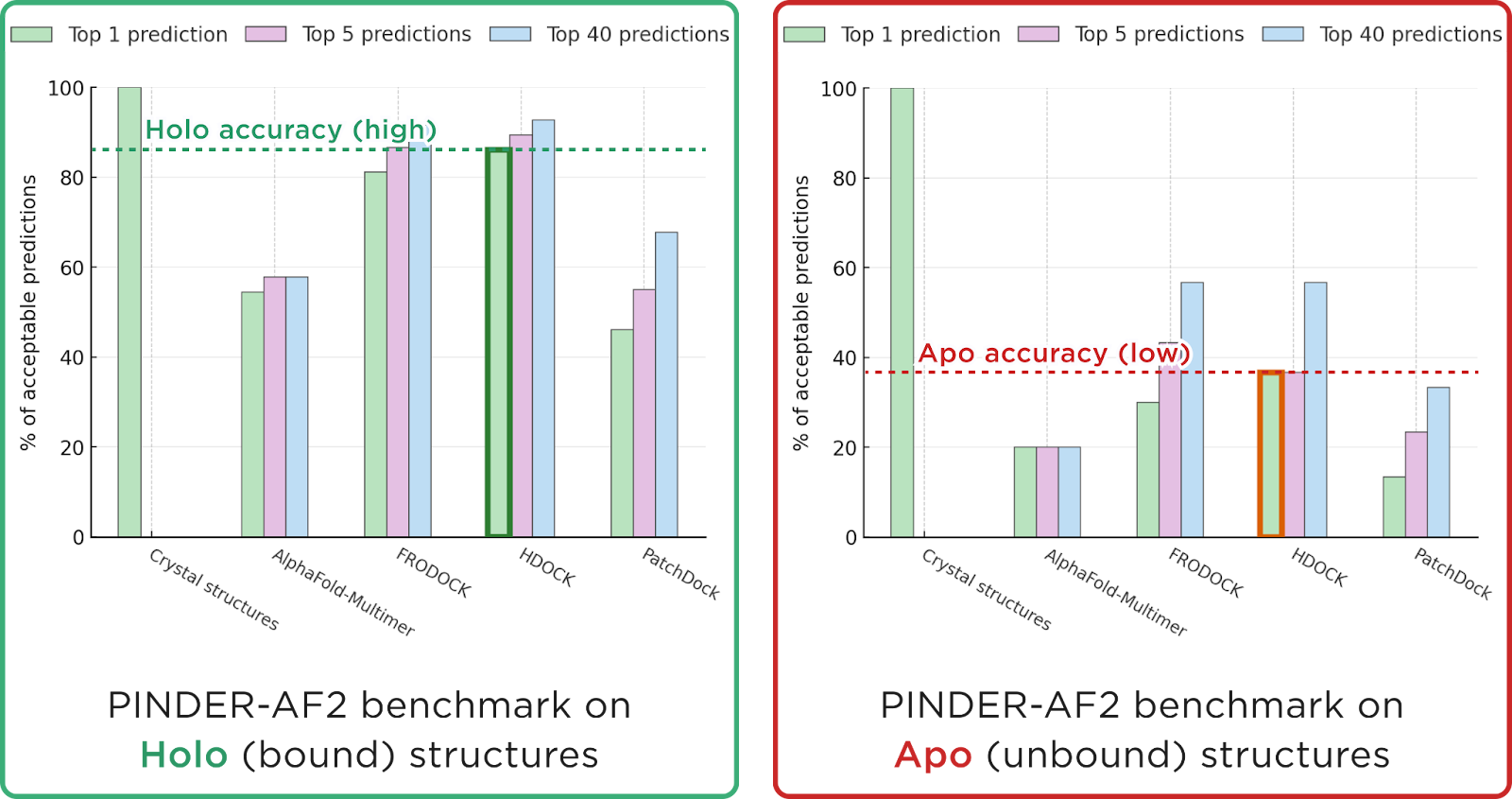
Co-folding methods like AlphaFold-Multimer are designed to capture flexibility. In theory, this should be an advantage, but in practice co-folding still performs worse than rigid docking on the most objective benchmark we have, PINDER.
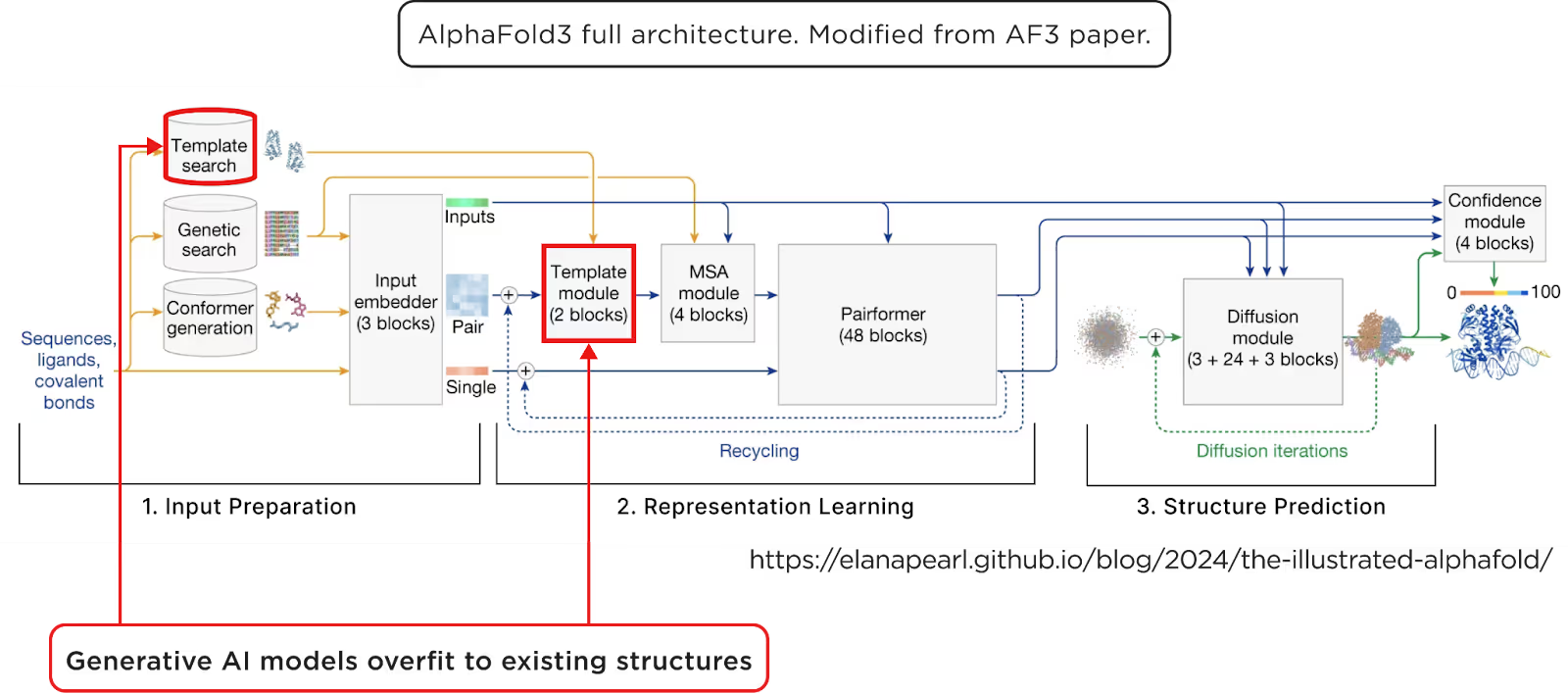
AI co-folding depends heavily on 3D complex templates. In the AlphaFold 3 architecture diagram, you can see the explicit usage of 3D structure templates during inference. But templates are rare, only ~1% of known PPIs. Moreover, more than 90% of these templates are stable, soluble complexes. Real biological PPIs are often transient, disordered, or membrane-bound. As a result, the training data is biased and scarce, and predictions suffer.
On the other hand, protein–protein docking appears to meet its accuracy limits. Induced-fit docking continues to underperform, while rigid docking depends heavily on the input structures, as detailed in our recent article. In short: rigid docking works well with Holo (bound) protein conformations as input and much worse with Apo (unbound) conformations.
Proposed Solutions
The first solution discussed at Promega was AI-driven conformational sampling for rigid docking, the very thing described in our article, so you might already know this approach didn’t work. Check for details.
Next there were details of Receptor.AI’s recent development: template-agnostic PPI prediction model, DeepTAG. Instead of docking proteins “as is”, it first predicts interaction hot spots, the local regions on protein surfaces most likely to drive binding. DeepTAG then matches these hot spots to assemble and evaluate candidate poses. The final step — complex relaxation via molecular dynamics simulation.
The hot-spot prediction module also requires training data, and the key insight is: intra-protein interactions follow the same fundamental physical rules as PPIs, so we can use them for training as well.
By extracting hot spots from both PPI templates and intra-protein contacts, we dramatically expand the dataset. On average, each protein yields 3 to 4 hot spots. With about 240,000 structures in the PDB, that translates to nearly 1 million hot spots available for training.
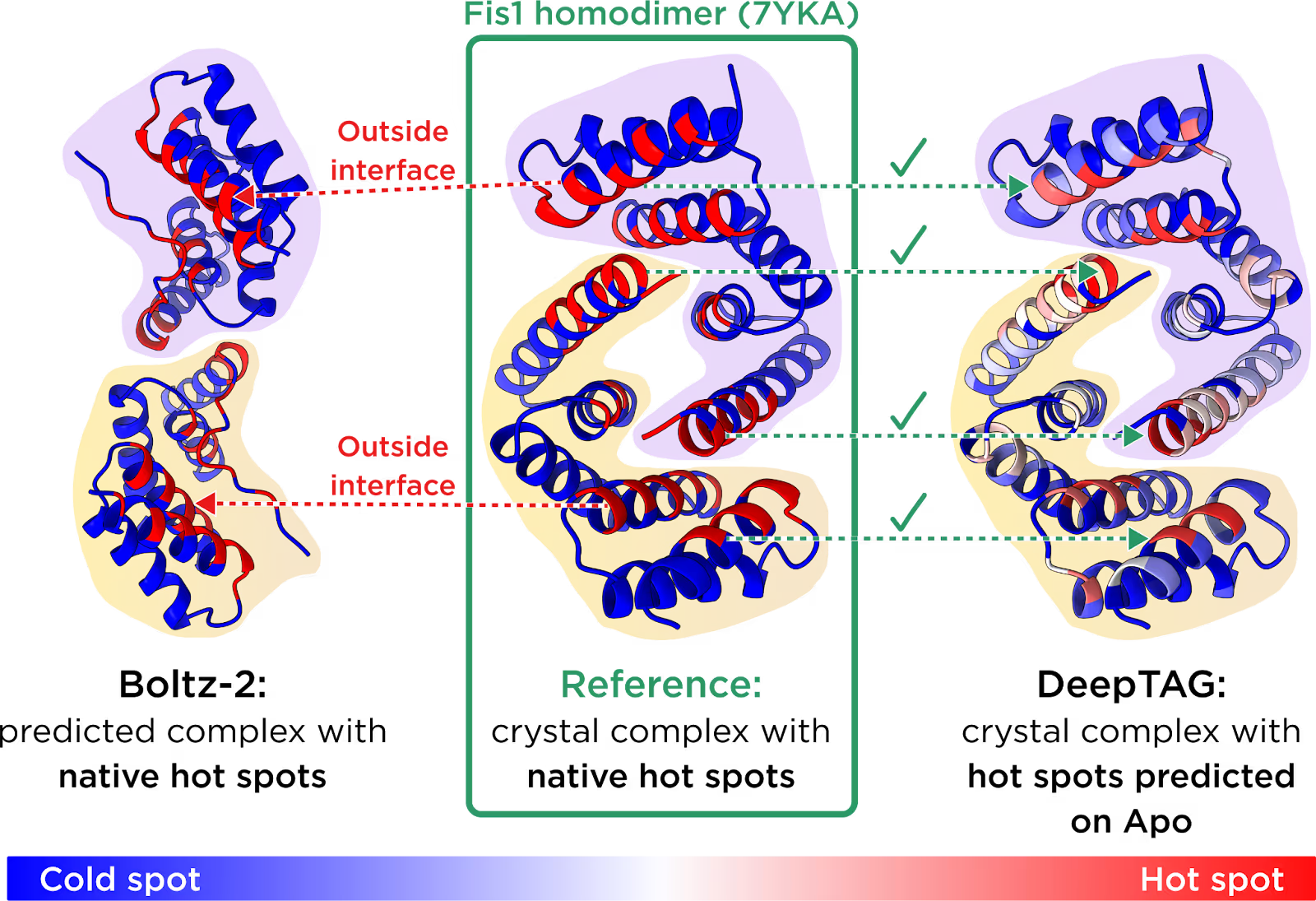
The hot spot prediction module was tested ahead of the Promega TPD Symposium. Here, we compare DeepTAG with Boltz-2 on the Fis1 homodimer. In the middle is the crystal structure with native hot spots highlighted. Left: the Boltz-2-predicted complex with the same native hot spots highlighted, showing that they do not interact in the predicted pose. Right: the reference crystal structure overlaid with hot spots predicted by DeepTAG on the unbound Fis1 conformation, closely resembling the native hot spots.
Conclusion
Although this technology is still in development, the potential is clear. Our approach can help address PPI data scarcity and model generalizability. AI delivers when applied at the right step in the workflow.