A structured approach to differentiating highly similar protein variants in drug discovery
Redefining Precision: Introducing Receptor.AI’s 5-Level Compound Selectivity Platform
A structured approach to differentiating highly similar protein variants in drug discovery
Announcement


Full Text
The ability to discriminate between several highly similar protein targets is crucial for modern precision and individualised medicine. Modern drugs are expected to be laser-focused on disease-related protein variants, which are specific to a particular tumour, tissue, cell type or the specific patient genotype while having no adverse off-target effects.
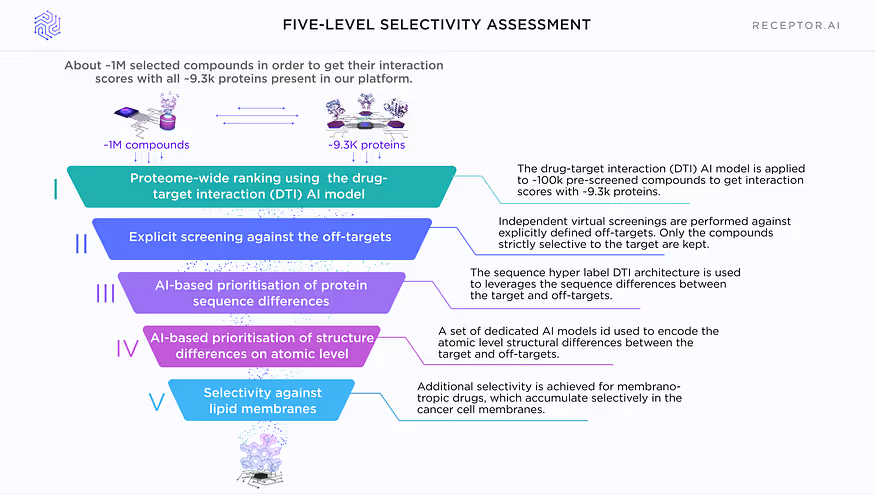
In order to achieve the goal of ultra-selectivity to similar protein variants, we created a unique set of technologies with five levels of selectivity prediction for the candidate compounds. The prediction accuracy increases on each level, so they are applied consecutively in the pipeline in concert with decreasing number of candidate compounds on each level.
This stack of technologies is incorporated into the Receptor.AI drug discovery platform and is available for any protein for which target and off-target variants could be determined.
Our platform is able to design ultra-selective small molecules for active and allosteric sites alike and operates even for the most challenging targets which lack known ligands or have poorly resolved structures.
In order to test our platform, we selected subsets of highly similar proteins from two most popular families of drug targets: Janus tyrosine kinases (JAKs) and kinases of Fibroblast growth factor receptors (FGFRs). These proteins are involved in a multitude of diseases, from cancer to cardiovascular diseases, inflammation and metabolic disorders. There is a large number of known ligands with reliable activity data for these proteins, which allows us to perform comprehensive and unbiased benchmarking of our technologies.
Five levels of selectivity prediction in a nutshell
Level 1: proteome-wide ranking
After initial virtual screening, the drug-target interaction AI model is applied to about ~100K selected compounds in order to get their interaction scores with all ~9.3k proteins present in our platform. The selectivity rank is computed, which is the number of proteins that are more likely to interact with a given compound than the target protein.
Level 2: explicit screening against off-targets
Independent virtual screenings against all explicitly defined off-target protein variants are performed. All compounds, which are selective to any of the off-targets, are discarded.
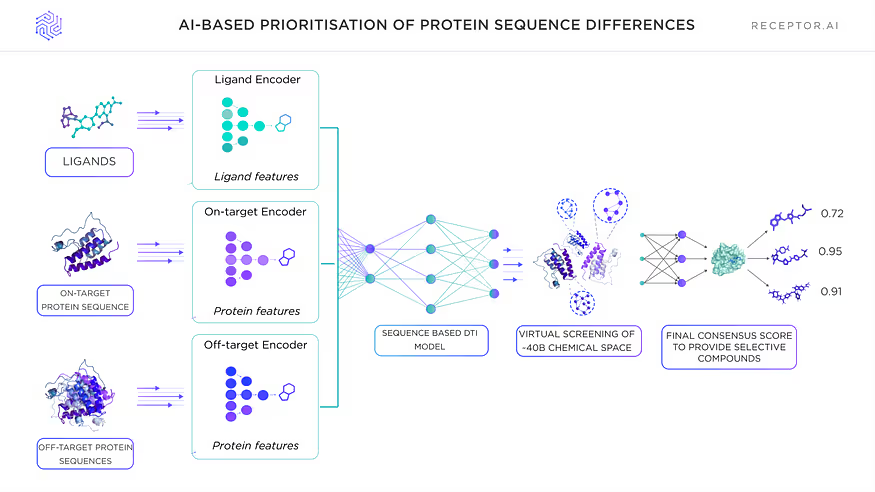
Level 3: AI-based prioritisation of protein sequence differences
A dedicated AI model based on sequence super-label architecture leverages and prioritises the differences between highly similar proteins on the level of protein sequences.
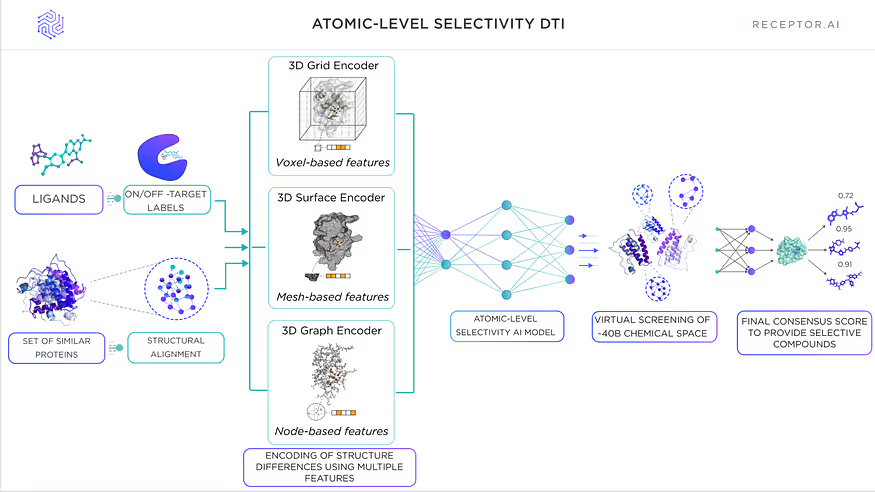
Level 4: AI-based prioritisation of protein structure differences on an atomic level
A set of dedicated AI models prioritises differences between highly similar proteins on the atomic level:
- Structure super-label DTI architecture
- Voxel-based selectivity model
- Enhanced AI-assisted docking
- Morphing transformer model
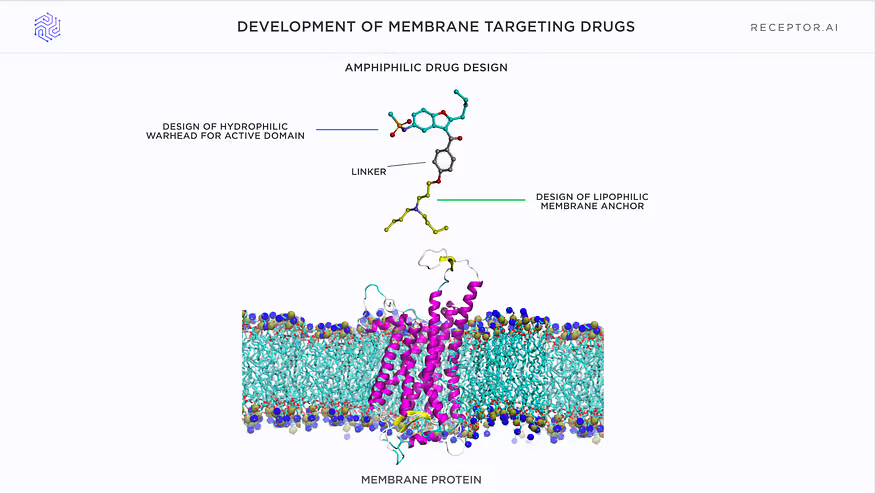
Level 5: Selectivity against lipid membranes (for cancer-related membrane targets)
Additional selectivity is achieved by designing candidate compounds as membranotropic drugs, which could accumulate selectively in the cancer cell membranes and be taken up preferentially by them. A unique AI model of drug-membrane interaction is used to search for such compounds.
The proof of the principle for such targeting was recently published.
Benchmarks
Target characterisation
The selectivity of compounds was determined separately against JAKs — JAK1, JAK2, JAK3, TYK2 and against FGFRs — FGFR1, FGFR2, FGFR3, FGFR4.
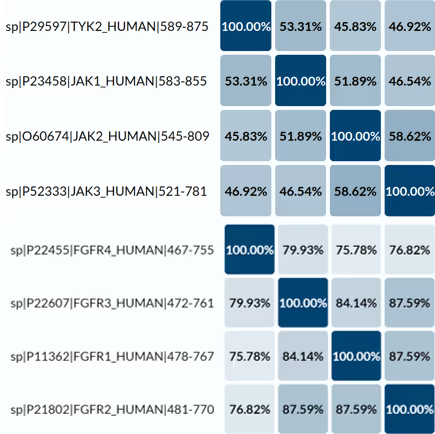
The overall sequence identity between the family members is rather small in the case of JAKs (40–60%) and much higher in the case of FGFRs (75–90%) (Fig. 1).
Despite these differences, both families could be characterized as highly similar proteins when comparing their functional binding pockets.
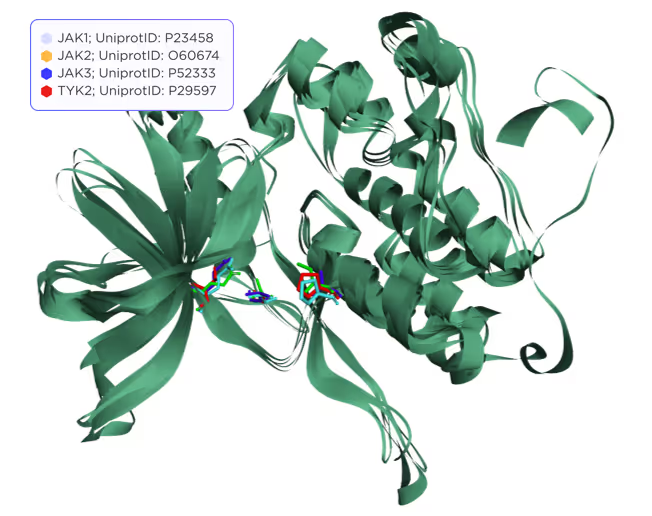
The active site of JAKs contains 15 functionally important residues, but only 3 of them are variable, while the rest is either identical or highly conservative. These residues are shown in Fig. 2 and 3.

In the case of FGFRs, the predicted binding pocket is rather large and contains about ~50 residues altogether, but only 2 of them are variable, while the rest are either identical or highly conservative (Fig. 4–5).

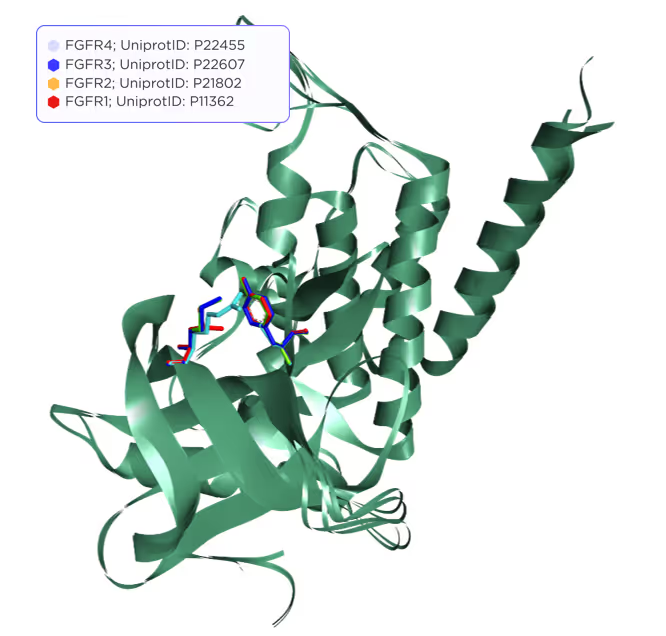
Our selectivity prediction technique emphasises the differences in a few variable residues automatically based on sequence and structural similarity between target and off-target proteins.
Compound dataset used in benchmark
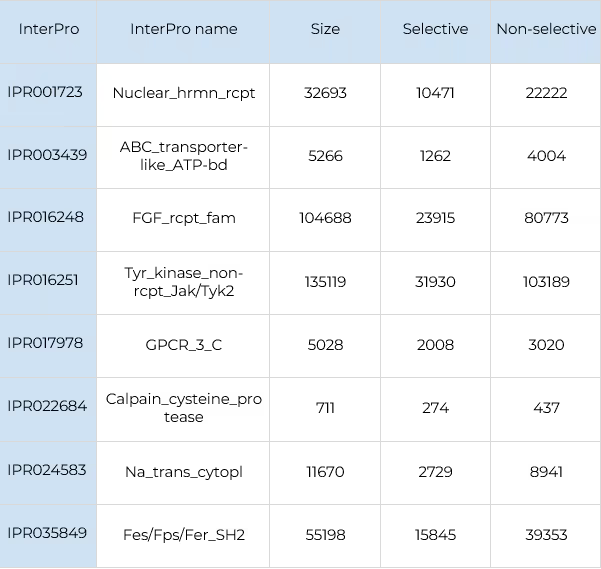
6830 compounds with known activity against the JAKs family and 4016 against FGFRs were taken from the ChEMBL database. All these compounds have activity against at least two kinases from the corresponding family.
The number of selective/non-selective compounds for each kinase according to this criterion in available experimental data is shown in Table 1.
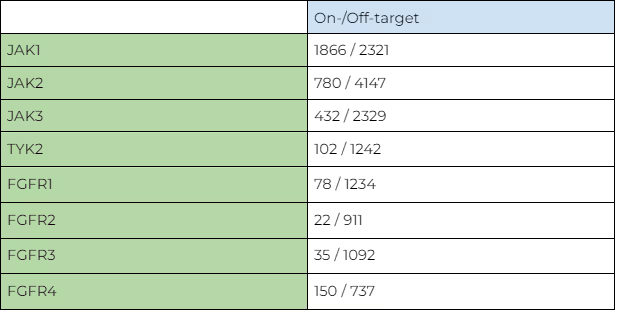
It is clearly seen that the dataset is significantly skewed. The largest number of compounds is found for JAK1, which is the most commonly used as a primary drug target in its family. This creates an inevitable bias towards JAK1 in the AI model. As it is shown in the results section below, the best results are indeed obtained for JAK1 due to the larger number of compounds available for training, while the results for JAK2, JAK3 and TYK2 are expectably worse because of insufficient training data.
For FGFRs the number of selective compounds is much smaller in general. The best coverage is observed for FGFR4, followed by FGFR1. FGRF1 and FGFR2 have very few selective compounds, which are barely enough for reliable benchmarking. However, even on such low-quality datasets, our models are providing good performance and consistent results, as it is evident from the results below.
In order to establish a reliable measure of compound selectivity, we plotted the ratio of the number of selective to non-selective compounds as a function of their experimental activity ratio (Figure 6).
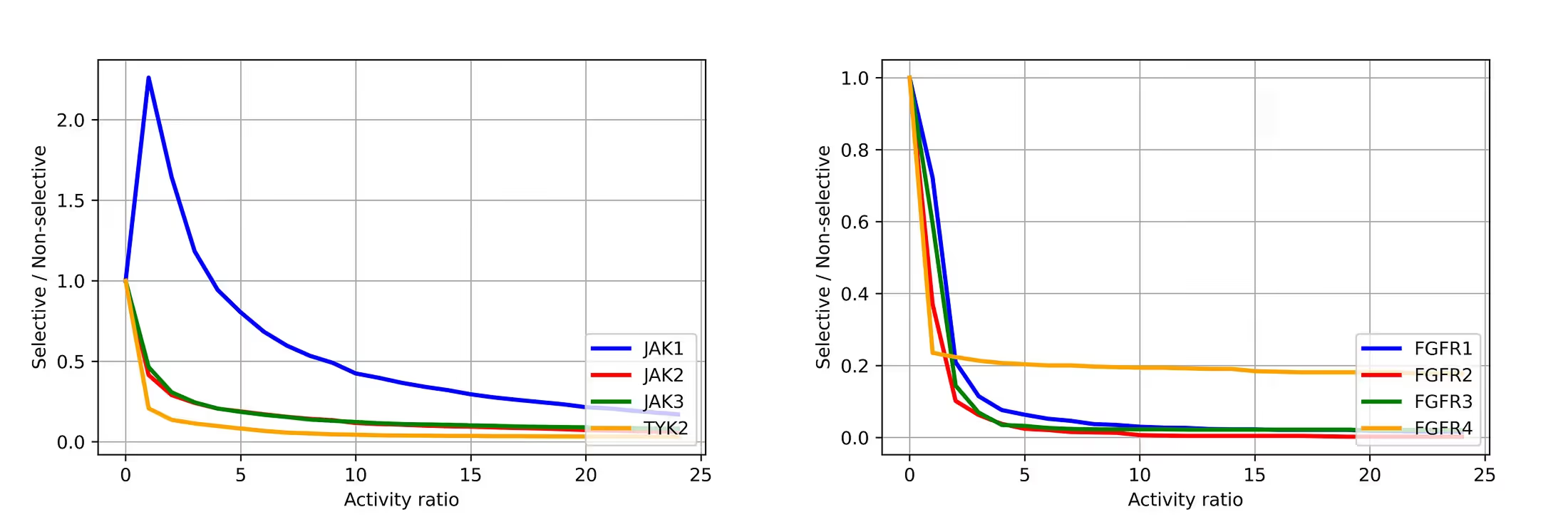
For FGFRs, the number of selective compounds becomes almost constant, starting from an activity ratio of 5. For JAKs, the general picture is similar except for JAK1, which shows a more gradual decrease. In general, an activity ratio of ~5 is a good threshold for establishing the compound’s selectivity for both kinase families.
Level 1: Drug-target interaction proteome-wide rank
Benchmarking procedure
A series of pairwise comparisons were performed when each kinase in a family was set as a target and all the rest as off-targets. A compound that is >= 5 times more active on the target kinase than on the off-target one was considered selective to the target.
The consensus scores of all compounds were computed using the Receptor.AI SaaS platform, setting each kinase consecutively as a target and the other one’s pair as off-targets. The pairwise differences between consensus scores were evaluated. If the difference is greater than the established cutoff, it indicates that the compound is selective and vice versa. This is a classical binary classification task, and accordingly, it is evaluated by the standard metrics for such problems. The main metrics are Matthews correlation and the F1 score, but a number of secondary statistical metrics were also computed. The metrics were computed for each pair separately. Then the metrics for all pairs were averaged, and the cumulative plots were built for each family.
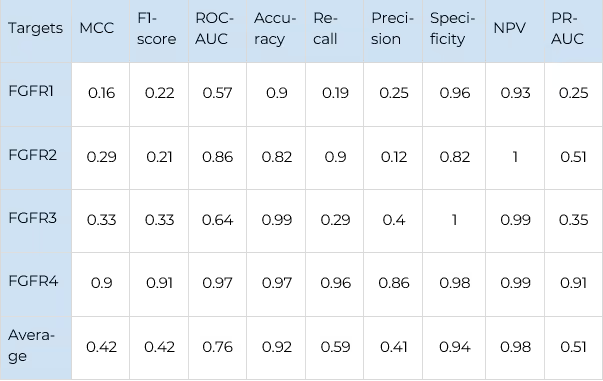
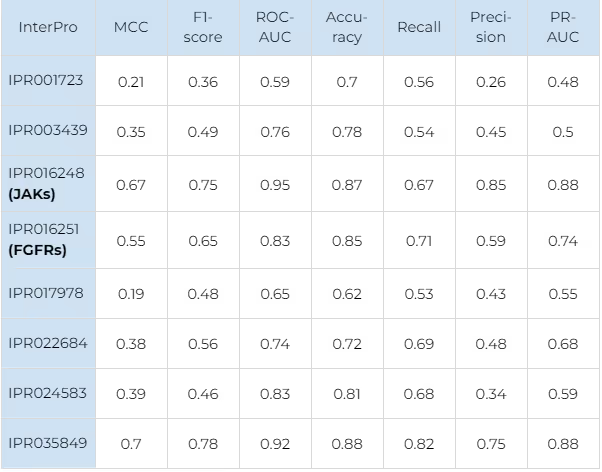
The main performance metrics of selectivity prediction are shown in Table 2 (for JAKs) and Table 3 (for FGFRs). The Receiver Operator Characteristic curves for the selectivity prediction (averaged for all kinases in each family) are shown in figure 7.
Results
Main performance metrics
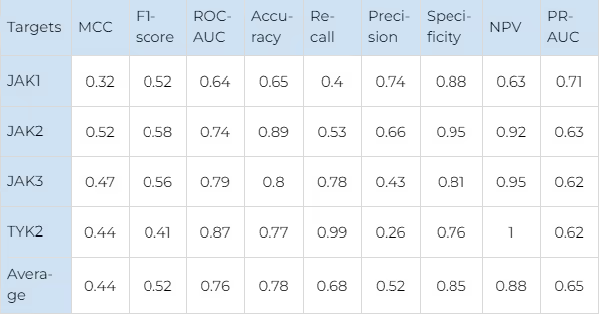
For JAKs the most robust results are obtained for JAK1 due to the largest number of data points available for this protein. For other JAKs, our model showed a worse distinction of false positives (precision values are worse) but a better percentage of correctly predicted selective compounds (recall values are better). It is remarkable that for TYK2, the model recognises almost all known selective ligands with an outstanding 0.99 recall value.
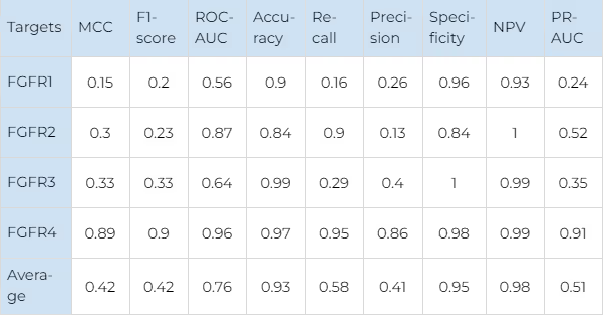
For FGFR2 the similar trend is observed. The smallest number of selective compounds is available, and almost all of them are predicted as such (recall — 0.9). However, the model predicts a significant number of false positives (precision — 0.13). For FGFR3, a comparable number of selective compounds is available, but the recall value is much worse (0.29), while the precision is much better (0.4). This indicates that although the model behaves differently on the targets with such a small number of data points, it still manages to balance the performance metrics in a consistent and predictable way.
Receiver Operator Characteristics and Enrichment Plots
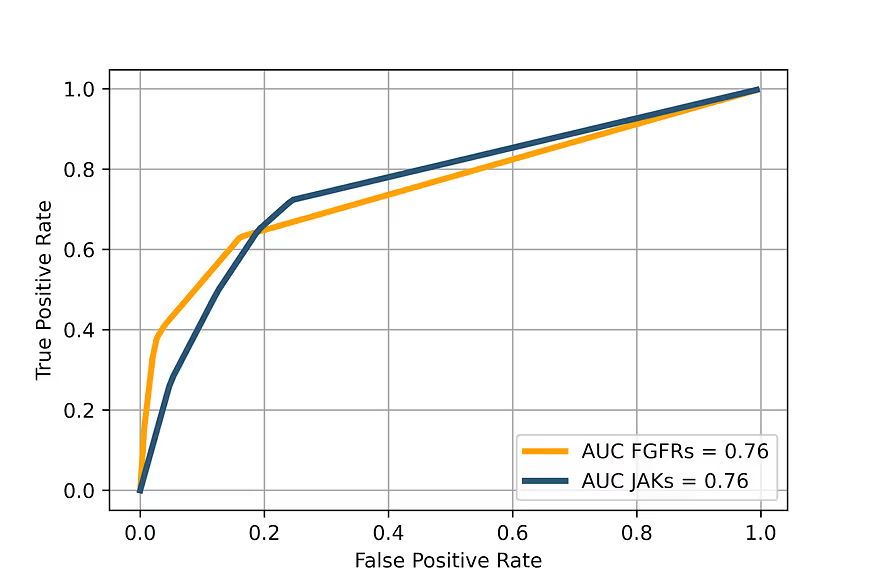
Our model provides very good average characteristics in distinguishing true positives from false positives. The best effectiveness is achieved for the top 20% of samples in the case of JAKs and for the top 10% for FGFRs. The general AUC is 0.76 in both cases, which is a very good value taking into account the data quality.
Comments on performance metrics consistency
The data for FGFRs is, in general, less abundant. FGFR2 and FGFR3 have only 22 and 35 selective compounds, respectively, which is on the lower boundary of the dataset size, which could be used for reliable benchmarking for this type of model. As it is evident from the results, this lack of data leads to some fluctuations in performance metrics, which is an expected behaviour.
For FDFRs, where very few selective ligands are present, each false negative (selective ligand, which was not found by our model) has more influence on the major metrics (F1 and MCC) than each false positive (non-selective ligand is incorrectly recognised as selective). That is why secondary metrics, such as recall and specificity, should also be used to evaluate the performance.
Specificity is the ratio of prediction of non-selective compounds, while recall is the ratio of prediction of selective compounds. Specificity values are very high for all proteins (around 0.9). This is very important for the virtual screening tasks because this means that the model correctly filters out the vast majority of non-selective compounds and doesn’t pollute the results with false positives. This makes subsequent experimental validation more effective and less costly.
The average recall values vary between 0.68 for JAKs to 0.58 for FGFRs. Both values are very good, taking into account the limited size of the dataset and the small number of positive samples.
Accuracy value provides a general ratio of correctly found selective and non-selective compounds to the total number of compounds. Accuracy values are also very good for all tested protein pairs, which confirms the general robustness of our approach.
Conclusions
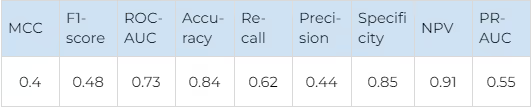
There is a good overall performance and a good balance between false positives and false negatives for both kinase families. The kinases in both JAK and FGFR families are very similar structurally, so such results confirm that our technique could discriminate between highly similar proteins effectively. The average metrics for both kinase families are shown in Table 4.
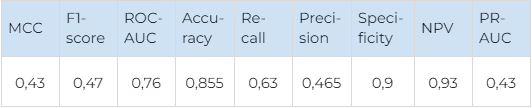
On average, the system predicts true selective compounds quite well (Recall 0.63) and discriminates non-selective compounds perfectly (NPV 0.9, Accuracy 0.855). That is, if a compound is non-selective, it is reliably identified and removed from further evaluation. 63% of the selective compounds are identified correctly. When applying the error of Accuracy, it turns out that 47% of compounds, which would be passed to experimental validation in a hypothetical experiment, are selective, which is an excellent ratio.
It is necessary to emphasise that the selectivity benchmark presented here is inevitably limited by the quality of testing dataset of compounds with known selectivity against different kinases. Particularly, there is pronounced bias in the number of available compounds for JAK1 in comparison to other kinases, which is caused by the popularity of this target protein. It is also evident that experimental activity estimates often differ in different pairs of proteins, which makes direct comparisons of the pairs less reliable. Taking into account these issues, the selectivity prediction shown by our technique could be qualified as very good.
Level 2: Explicit virtual screening against multiple off-targets
Level 2 of selectivity prediction is full screening against explicitly defined off-targets with exclusion of all compounds, which are selective against off-targets according to the difference between corresponding consensus scores.
Benchmarking procedure
Test test sets were the same as in Level 1 — JAK and FGFR proteins. Benchmarking procedure was also the same. Instead of DTI Rank, we predicted selectivity based on the difference between consensus scores of the target and off-targets.
Results
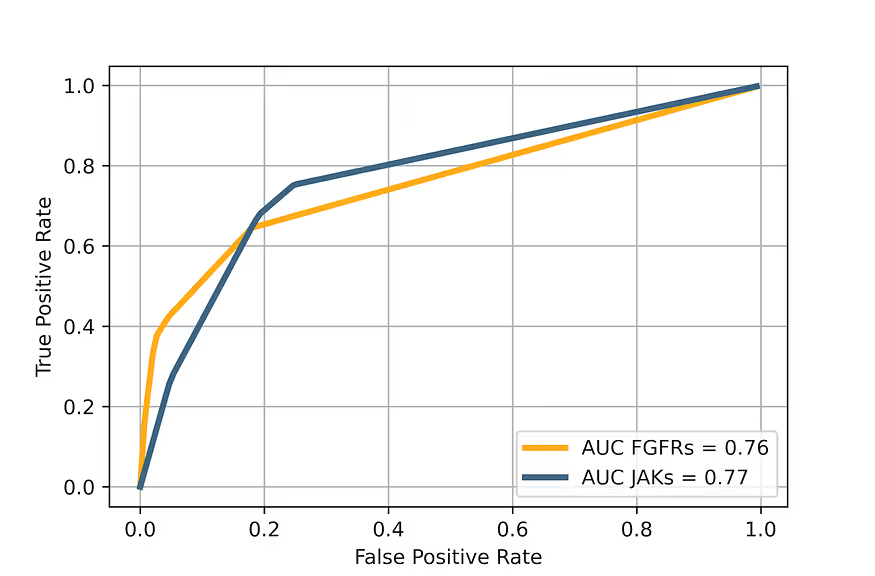
The main performance metrics of selectivity prediction are shown in Table 5 (for JAKs) and Table 6 (for FGFRs). The Receiver Operator Characteristic curves for the selectivity prediction (averaged for all kinases in each family) are shown in figure 8.
Main performance metrics
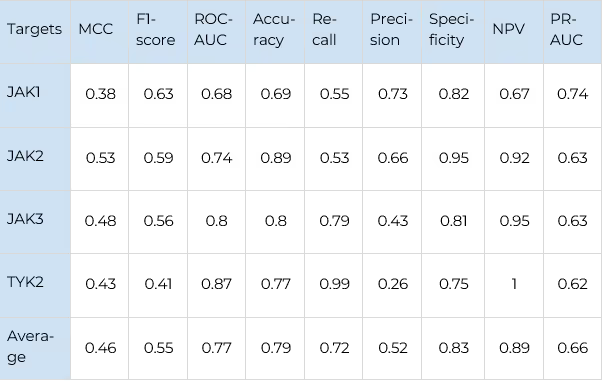
Receiver Operator Characteristics and Enrichment Plots
Conclusions
Results on Level 2 are similar to results on Level 1, but a little better.
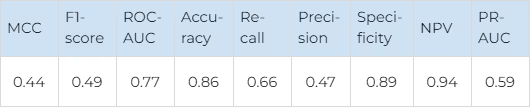
There is a good overall performance and a good balance between false positives and false negatives for both kinase families. The kinases in both JAK and FGFR families are very similar structurally, so such results confirm that our technique could discriminate between highly similar proteins effectively. The average metrics for both kinase families are shown in Table 7.
Level 3: Selectivity prediction based on protein sequence differences
Level 3 of selectivity prediction is implemented as virtual screening against the main target and its off-targets by the ESM Selectivity model, which operates at the level of protein sequences.
Model description
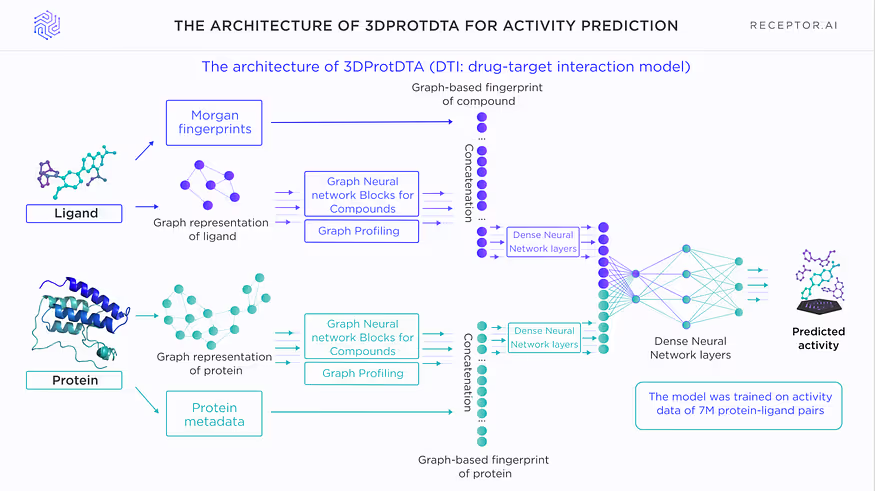
On this level we utilise the pocket-agnostic AI model for predicting selectivity of the ligands based on the ESM sequence encoding technology (https://github.com/facebookresearch/esm).
Model does not perform separate screenings against the target and off-targets. Instead of this it operates with the super-labels constructed from the ESM representation of the target, ESM representation of the given off-target and the fingerprint of the ligand of interest. The model outputs a selectivity score ranging from 0 to 1. The higher the score, the more likely the compound is selective.
The model was trained on all possible combinations of labelled triplets consisting of either the “target/off-target/selective ligand” or “target/off-target/non-selective ligand”. The compounds with known biological activity against the protein targets of all organisms were used for training.
Benchmarking procedure
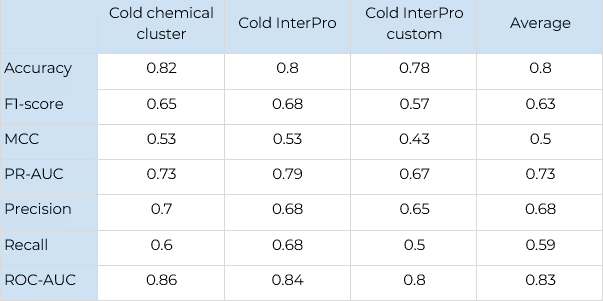
Testing of the model was performed on three test sets: “cold chemical cluster”, “cold InterPro” and “cold InterPro custom”.
- The “cold chemical cluster” set consists of 200 clusters of compounds from the ChEMBL database, which were not used for model training. The clusters were computed using k-means algorithm operating on Tanimoto distance between the Morgan 1024 bit fingerprints of radius 3.
- “Cold InterPro” set consists of 10 randomly chosen protein families from the InterPro protein functional classification, which were explicitly excluded from the model training.
- “Cold InterPro custom” set is the same as the previous one, but the list of protein families was prepared customly.
Results
The main performance metrics of selectivity prediction are shown in Table 8 and detailed information about experiment on Cold InterPro custom — in Tables 9 and 10.
Conclusions
There is a good performance of Level 3 selectivity. The tests show robustness of this technique because it performs well with either customly selected or randomly chosen protein families.
Performance metrics of selectivity prediction for JAK and FGFR families are especially good (IPR016248 and IPR016251 in Table 6 respectively).
Level 4: Selectivity prediction based on protein structure differences
* The model is in the alpha version and is currently under active development.
Level 4 of selectivity prediction is implemented as virtual screening against the main target and its off-targets by the structure-based selectivity prediction model, which operates at the atomic level of protein-ligand connectivity of ligands in the binding pocket.
Model description
On this level we utilise the structure-based pocket-oriented AI model for predicting selectivity of the ligands. The idea and architecture of the model are inspired by the CLIP neural network architecture developed by OpenAI. In our case instead of constructing the joined multimodal embedding space for images and text we build a common embedding space for the binding pocket atoms and the corresponding ligand atoms. The model is optimised to produce embeddings for pocket and ligand atoms that are similar (high cosine similarity between embeddings) for the pairs of interacting pocket and ligand atoms, while being strongly dissimilar (low cosine similarity) for non-interacting pairs.
The model performs separate screenings against the target and off-targets with exclusion of all compounds, which are selective against off-targets according to the difference between corresponding scores.
The model was trained on all 3D complexes of the “target/ligand” pairs from PDB. The compounds with known biological activity against the protein targets of all organisms were used for training.
Benchmarking procedure
Test test sets were the same as in Level 1 (JAK and FGFR protein families). Benchmarking procedure was also the same.
Results
The average performance metrics of selectivity prediction are shown in Table 11 (for JAKs and FGFRs).
Comparison of different selectivity levels for JAKs and FGFRs
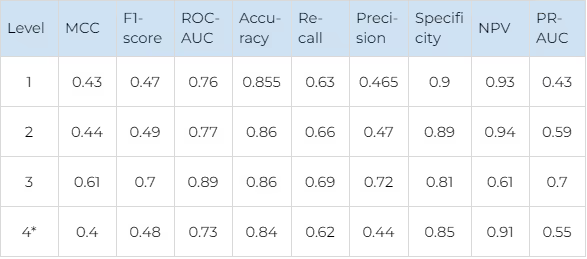
*The model is in the alpha version and is currently under active development.
Level 5: Selectivity to the membrane composition for the membrane targets
This level of selectivity is reserved for the membrane proteins related to cancer.
The plasma membranes of cancer cells are distinct from their normal counterparts. Additional selectivity is achieved by designing candidate compounds as membranotropic drugs, which could accumulate selectively in the cancer cell membranes and be taken up preferentially by them. Interaction of the candidate compounds with the cancer cell membrane is ensured by applying a unique AI model of drug-membrane interaction. The proof of the principle for such targeting is recently published by the CSO of Receptor.AI.
Conclusion
Following extensive testing, the 5-level compound selectivity platform developed by Receptor.AI has demonstrated promising outcomes. The platform’s unique set of technologies, designed to predict ultra-selectivity to similar protein variants, performed with increasing accuracy across each level. Tested on subsets of highly similar proteins from two of the most common families of drug targets, Janus tyrosine kinases (JAKs) and kinases of Fibroblast growth factor receptors (FGFRs), the platform showed its robustness and precision. These results highlight the platform’s potential in designing ultra-selective small molecules for both active and allosteric sites, even for targets with no known ligands or poorly resolved structures. This successful testing reaffirms the platform’s readiness to contribute significantly to the field of precision and individualised medicine.
In the near future, we will implement a novel model that utilises generative AI to account for the pharmacophore features of binding pockets of targets and off-targets, with these features being gathered from trajectories derived from comprehensive molecular dynamics simulations.