AI predicts results of complex chemical reactions with 90% accuracy
The AI that speaks the language of chemical reactions natively
AI predicts results of complex chemical reactions with 90% accuracy
Announcement


Full Text
The problem which is kind of solved but not really
The history of rational drug discovery is a constant drift from experiments in the wet lab towards in silico simulations. This trend is easily explained by much lower cost and much higher speed of simulations in comparison to even the most efficient and automated experimental procedures.
In the early days of the high-throughput screening, it was the initial step of filtering the chemical universe in search of promising active compounds. Nowadays, the primary filtering is done computationally, and the real-world screening is performed on the subset of carefully pre-selected compounds. This optimized the process and dramatically increased the success rate in the early stages of the drug discovery pipeline but didn’t eliminate the need for experimental validation.
The validation of computational hit compounds starts from the obvious, routine, and thus often overlooked stage — their chemical synthesis. Modern in silico screening techniques, especially those based on AI models, prioritize the molecules, which look “easily producible” or even available right off the shelf from the commercial chemical suppliers. Big players on this market propose the libraries of the billions of compounds, which could be produced in a few days and shipped worldwide. That is why the problem of chemical synthesis in the drug discovery pipeline is often considered “solved” and purely technical.
However, it is not all roses. First, computational molecular generators and subsequent virtual screening may quickly produce exotic chemical species, which have never been seen before. The possibility of synthesizing such compounds is crucial for the early-stage experimental validation and the overall feasibility as drug candidates in the long run. Indeed, if the synthesis of even the most efficient drug is so complex and expensive that it hampers its commercial production, market perspectives become vague.
Second, the computational molecular generation and screening itself need reliable metrics of “synthesizability.” There is vast literature on this topic, but there is no silver bullet yet. Most of the available techniques are structure-based and rely on analysis of the chemical groups in the molecule in terms of their synthetic accessibility. Although these techniques are rather efficient and robust, the problem is far from being solved.
Speaking the language of chemistry
The problem of synthesizability is complex and is based on an overwhelming amount of data; thus it is tempting to deal with it using machine learning approaches. The classical way of thinking is the following. Let’s compose a huge training dataset containing the molecules and their “synthesizability scores” (whatever it means). After that one trains an AI model, which relates the chemical structure of the compound with its score and allows predicting this score for any new molecule.
Such AI models work, but they have one significant drawback — they do not follow real chemical reactions. The AI model is not aware of actual synthesis pathways and their relative complexity but rather assesses only the final result of these complex processes.
A fundamentally different approach was proposed recently. The idea is to teach the neural network to speak the chemical language literally.
Natural human languages consist of words combined using grammar rules and semantics. These rules are complex, but there is an architecture of the artificial neural networks which excels in the natural language processing — the transformers. The transformers are deep learning models that adopt the mechanism of attention and differential weighting of each part of the input data in terms of their significance. These models transform the input sequence of tokens into the output sequence, which is ideal for language processing and generation tasks.
Recent years’ most famous transformer models are from the GPT family, which gained a lot of hype in mass media because of their ability to produce impressively realistic texts. However, transformers are not limited to the natural language. For example, they allowed the AlphaStar AI to defeat top professional Starcraft players. These models also excel in different protein-related biological problems by considering the primary sequence as an input text, which translates into various properties of the functional protein globule. The most famous protein-related transformer model is the AlphaFold2, which has recently de facto solved the protein structure prediction problem.
Baby steps: the alphabet and the chemical words
So, what language should the model learn to follow the reactions of the chemical synthesis? It is not obvious what are the “letters” and the “words” of the chemical structures. In contrast to the texts in human languages or protein sequences, the networks of molecular bonds are not linear. Chemical groups could not only attach to each other, but also intercalate and cross-link in a complex way. The branched molecular graph is fundamentally different from the linear stream of input tokens, which transformers are so efficient in operating with.
The rescue is SMILES — the simplified molecular-input line-entry system (SMILES), which describes any chemical structure as a string of symbols encoding the atoms, bonds, branching, cycles, aromaticity, stereoisomers and even isotopes.
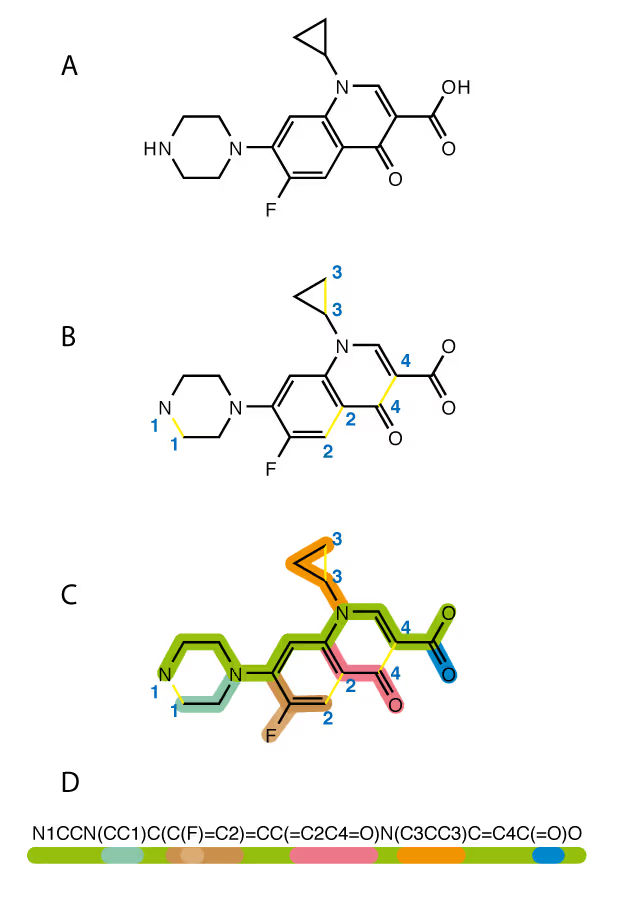
Each symbol of SMILES string is considered as a separate “word” (an input token). The neural network learns which combinations of such words may appear and in which contextual surrounding inside the molecule. Each molecule could be considered as a grammatically correct sentence. The model is agnostic about real physical meaning of the tokens. It just learns the “language” of the SMILES strings.
Making meaningful texts: chemical reactions
The next step in mastering the chemical language is combining the sentences into the meaningful “sayings”. This is done by learning the rules of chemical reactions. Each reaction takes the list of reactants and produces the list of products. In terms of our language metaphor, one paragraph of chemical text is being transformed into the other paragraph following rather complex rules. We can’t formalize these rules, but we could learn by example by looking on large set of chemical reactions.
Although huge amount of reactions is possible, not all of them are useful. The training dataset should contain only “good” reactions, which are acceptable for commercial use. Very good, but somewhat not obvious, source of such reactions are registered patents. Indeed, nobody will ever base a patent on a useless reaction, which is hard to perform on practice.
The training dataset contained chemical reactions mentioned in US patents published between 1976 and 2016 divided these data into independent training and validation sets.
Increasing the vocabulary: data augmentation
The real human languages are full of synonyms. In order to speak and be understood, one needs just a few hundred of words, but much more is necessary to reach the level of the native speakers. Moreover, the same meaning could be expressed by a number of different sentences constructed from different words in different order.
In terms of SMILES chemical representation, this comes in the form of redundancy of SMILES strings. The same molecule could be described by a number of different SMILES strings, which are all chemically equivalent. Indeed, one can start enumerating atoms from different ends of the molecule and close the loops of aromatic cycles in a different sequence. In order to overcome such redundancy the so called canonical SMILES were introduced. Such representation is unique for any given molecule, but, to complicate the matters, the canonicalization algorithm itself is not standardized.
Anyway, the AI model, which speaks chemical language fluently, should understand different redundant SMILES sentences and produce the same results regardless of the “synonyms” used. To achieve this original training dataset was augmented with alternative representations of the SMILES strings, which significantly increased it size and gave the neural network a lot more data to learn on.
This approach is somewhat similar to the techniques used in computer vision AI models, where the neural network have to recognize particular object regardless of its size, color and orientation in space. In this case the model should understand the chemical text regardless of the SMILES variants used.
Passing the exam
The authors validated the model by a blind prediction of the reaction products using the validation dataset. The overall model performance was evaluated by the number of correctly predicted reaction products.
The AI models produce the list of predicted reaction products, which are ranked by their likeness to appear. Such probabilistic output means that even if the real product doesn’t coincide with the top-ranked output it can still be present as an output ranked second, third of tens. Thus the percentage of matches among top 10 predicted molecules for validation dataset was computed. The result looks fantastic:
- Top 1: 85.1%
- Top 2: 89.8%
- Top 3: 91.2%
- Top 5: 92.5%
- Top 10: 93.5%
This means that 85 out of 100 chemical reactions are predicted with perfect accuracy. If we take one of the top-10 ranked predictions, this number increases to 93 out of 100.
Usage and perspectives
The AI-based deep-learning neural networks, which simulates the chemical reactions with high accuracy, are immediately useful in a number of applications:
- A playground for organic chemists. The user can change reagents and reactants and immediately see the changes in the products, which are likely to occur.
- Confirmation of synthesizability. All reaction products generated by the model are priory synthesizable, and all the reagents needed for obtaining them are known.
- A playground for organic chemists. The user can change reagents and reactants and immediately see the changes in the products, which are likely to occur.
- Engine for combinatorial chemistry. Being provided with the list of possible reagents, the model can estimate the feasibility of all their combinations and predict the resulting set of products.
- “Reverse engineering” of reagents. Given the hit compound found in the course of the drug discovery project, the best synthesis pathway could be proposed.
The AI model described here is only one of the multiple innovative technologies which are used and improved in Receptor.AI.