
The Complexity Bias: Why ML Drug Discovery Models Sometimes Fail in Practice
Announcement


Full Text
Our recent results show that even the most advanced ML-generated protein conformations do not enhance rigid protein–protein docking, even when modern scoring functions are used to rank the docking results.
What we tested
Rigid protein–protein docking is still widely used for protein-protein complex assembly, and is accurate if the bound conformation is provided as input. However, it does not account for induced-fit, which is detrimental for accuracy, especially when it starts from unbound (Apo) structures.
We wanted to know: If we generate bound(Holo)-like protein conformations using modern ML methods and subject them to protein-protein docking, will we increase the rigid docking accuracy?
To find out, we ran AFSample2 and BioEmu on monomers from 30 protein–protein complexes.
These methods have gained attention for their ability to produce diverse structural ensembles using AlphaFold-like architectures. AFSample2 is widely used for sampling multiple conformations with minimal overhead, while BioEmu has been positioned as a high-performance alternative for capturing functional protein flexibility at scale. Both are considered among the most capable tools for ML-driven conformational sampling today, which is exactly why we chose to test them.
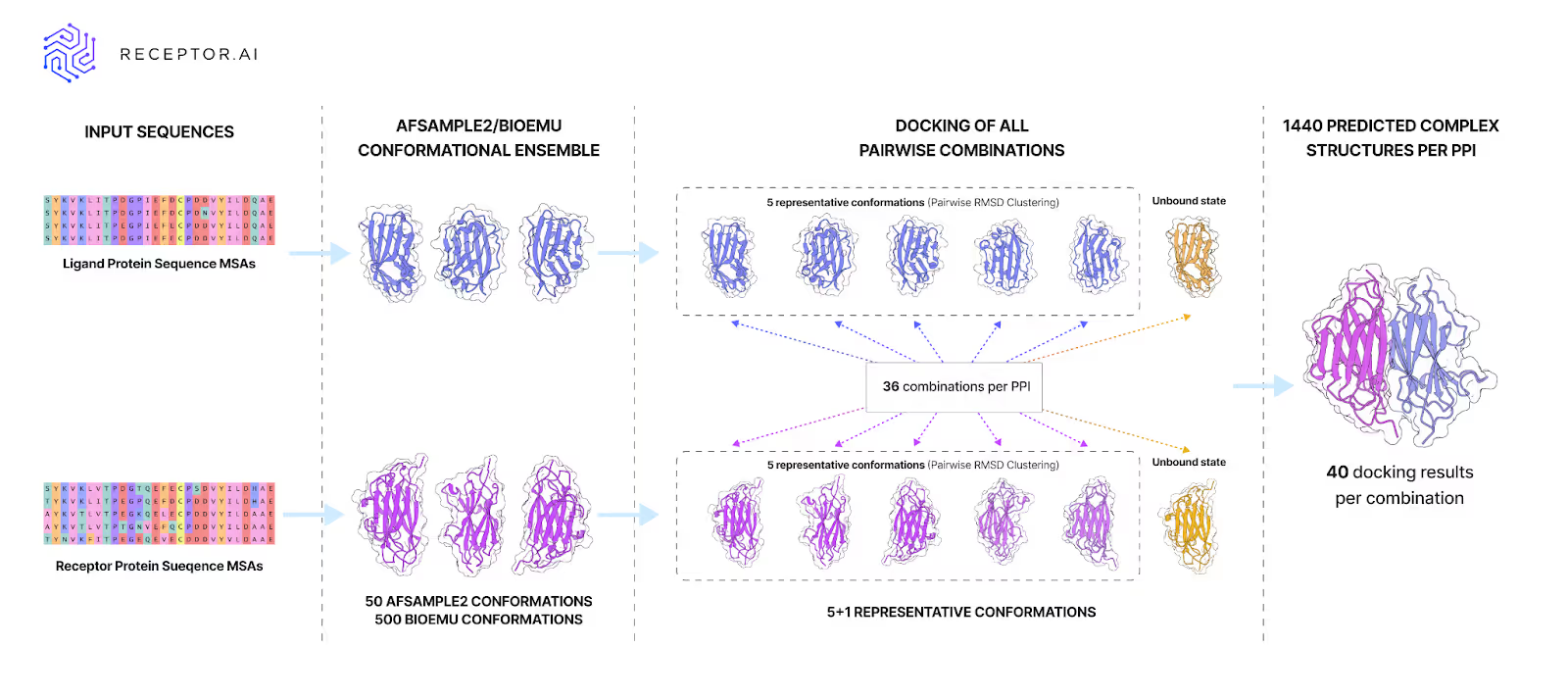
We used PINDER-AF2, the most accurate and representative benchmark, free from data leakages and thus suitable to evaluate how well methods generalize on unseen protein interactions. We docked the sampled structures and ranked results with five scoring functions to see if the most advanced approaches available today could prioritize native-like PPI structures.
What we found
AFSample2 occasionally produced promising conformations (closer to the Holo state), but the improvements were marginal and inconsistent.
BioEmu struggled throughout. Most of its sampled structures were even further from the Holo state than the original Apo form and some were completely unnatural. Increasing the number of sampled conformations didn’t help either. More data simply created more noise.
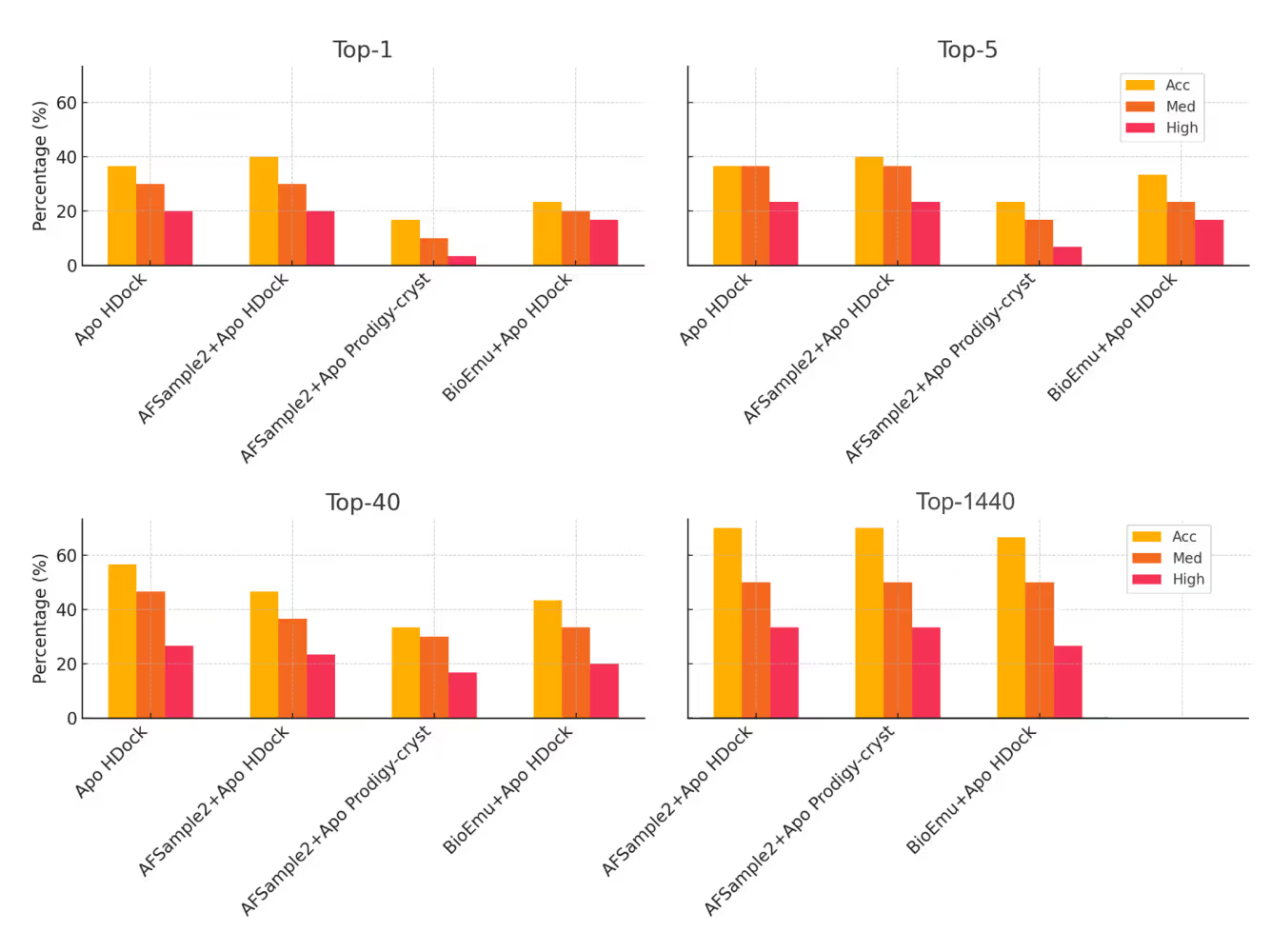
But even when promising structures were present, none of the five tested scoring functions, including HDOCK, PRODIGY-Cryst, HADDOCK3, dMaSIF, and PIsToN, could reliably pull them from the heap. In multiple cases, high-quality docked models were in the pool, but misranked so badly that they were even beyond the Top-40.
In short, generation fell short, but scoring proved to be the more decisive bottleneck.
So what does this tell us?
There’s a common bias in the field that once models reach a certain level of complexity, they can operate in simple workflows and still perform well. But that’s not what we saw.
Even state-of-the-art sampling methods struggled in production. The simpler the setup, the more honest the results, and right now, these models aren’t ready for standalone use. Models that look impressive in isolation can still fail under realistic conditions.
What’s our approach?
At Receptor.AI, we use a template-agnostic PPI prediction model, DeepTAG, designed to work from the bottom up.
Instead of docking the protein structures and relying on scoring to sort them out, we first predict interaction hot spots, the local regions on protein surfaces most likely to drive binding.
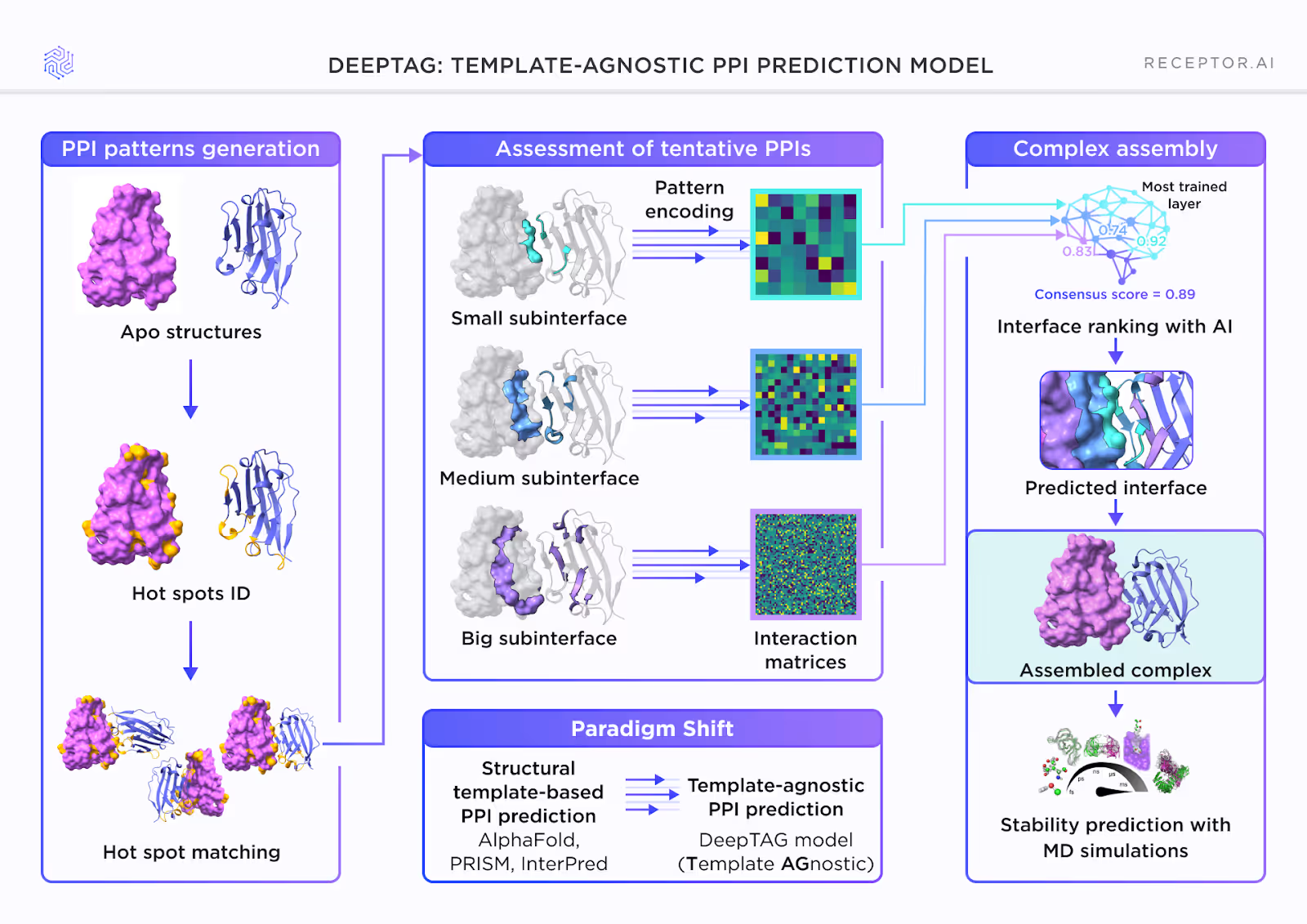
From these hot-spots, we assemble and evaluate candidate interfaces, scoring them using models trained to recognize meaningful structural features. Only then do we construct full complexes, using the best-scoring interfaces as anchors.
This bottom-up approach, from local signals to assembled structures, lets DeepTAG avoid the combinatorial noise of traditional docking and predict PPIs more efficiently and accurately.
Conclusion
For now, there’s still a gap between model complexity and practical robustness. But this isn’t a failure of AI. It’s a reminder that progress depends on how well we build around it.
Resources & Links
What is PINDER? The most modern, leakage-free docking benchmark.