The Intersection of Generative AI and Molecular Dynamics in Drug Discovery: Limitations and Opportunities
Announcement


Full Text
This post discusses how advanced AI methodologies are revolutionizing drug discovery, focusing on molecular dynamics (MD) simulations, conformational sampling, and drug-target interaction prediction. Drawing on our work at Receptor.AI, we highlight how Generative AI enhances drug candidate identification beyond traditional methods.
We also address challenges in applying ML to MD, such as data quality dependence, overfitting, and navigating kinetic barriers, sharing strategies we've successfully employed to overcome them.
We aim to foster dialogue and share expertise in AI-driven drug design. Researchers, drug discovery professionals, and medicinal chemists are invited to share insights or collaborate, open-source projects included.
Modern challenges
Molecular Dynamics (MD) simulations model the behavior of atoms in complex systems like proteins by calculating the positions and velocities of each atom over time. The inherent high dimensionality arises because each atom contributes three coordinates for position and three for velocity. This leads to a large computational workload in tracking their motions. For instance, simulating a protein with thousands of atoms involves millions of calculations per time step, making it computationally expensive, especially for capturing biologically relevant timescales. The most computationally demanding task is calculating non-bonded interactions, such as van der Waals forces and electrostatic interactions. The number of these pairwise interactions scales quadratically with the number of atoms. For example, a system with 10,000 atoms has approximately 50 million possible pairwise interactions. Although the use of cutoff distances and efficient algorithms significantly reduces the actual number of calculations per time step, calculating non-bonded interactions remains a primary bottleneck in simulation performance.
Furthermore, the challenge is exacerbated by the vast conformational space that proteins can explore. Proteins may adopt numerous stable and intermediate conformations, often trapped in local energy minima. These kinetic barriers, arising from intramolecular interactions like hydrogen bonds and hydrophobic effects, hinder transitions between states. Traditional MD simulations, with time steps in the femtosecond range, struggle to reach the millisecond or eric effects, and ligand binding pathways, enhancing ML models with a more extensive understanding of protein dynamics. Additionally, many proteins perform their functions through dynamic conformational changes, including allosteric regulation, induced fit substrate binding, and signal transduction. Static structures often miss these intermediate or alternative states crucial for understanding protein behavior. Integrating MD-derived features, such as RMSD, RMSF, solvent-accessible surface areas, principal component analysis, etc. allows for improved generalization and reduces overfitting in predictive models, making it a critical approach for capturing the intrinsic dynamic nature of proteins in computational studies.
However, in order to train high-performing ML models on MD, the data must effectively represent the real-world conformational ensemble. Obtaining such data remains challenging. A major issue in leveraging MD data for ML training is related to one of ML's core challenges: generalization beyond observed data.
ML models trained on MD datasets often struggle to generalize beyond the sampled conformational space, which may miss rare or high-energy conformations due to MD's inherently limited sampling. While AI models in general are sometimes criticized for their inability to extrapolate, particularly across energy barriers, this perspective overlooks some recent findings. For instance, the study "Learning in High Dimension Always Amounts to Extrapolations" by Facebook AI Research and New York University shows that in high-dimensional spaces, ML models almost always operate in an extrapolation regime, which does not necessarily hinder performance or generalization. However, this extrapolation ability depends on the quality of the training dataset and its curation for this particular purpose.
Curating datasets for rare features, like transient high-energy conformations, goes beyond simply increasing structural diversity. It involves ensuring that the data captures relevant physical phenomena governing molecular transitions, emphasizing the importance of quality over quantity in dataset design. To achieve strong performance in drug design, datasets must reflect these complexities.
Exploring Successful Cases
Despite obvious challenges, there are successful cases of direct generation of protein conformational ensembles using AI. For example, the IdpGAN model described by Janson et al. (2023) in “Direct generation of protein conformational ensembles via machine learning” is a generative adversarial network (GAN) designed to produce 3D conformations of intrinsically disordered proteins (IDPs) at a Cα coarse-grained level using MD training data. The architecture features a generator (G) and multiple discriminators (D), where G is based on a transformer architecture. The generator takes a latent sequence and amino acid information as input, outputting 3D coordinates of Cα atoms. Discriminators evaluate these generated conformations by comparing distance matrices against real MD samples, making the model invariant to transformations like rotation or translation. IdpGAN was trained on simulations of IDPs with varying lengths (20-200 residues) using residue-level coarse-grained and all-atom models.
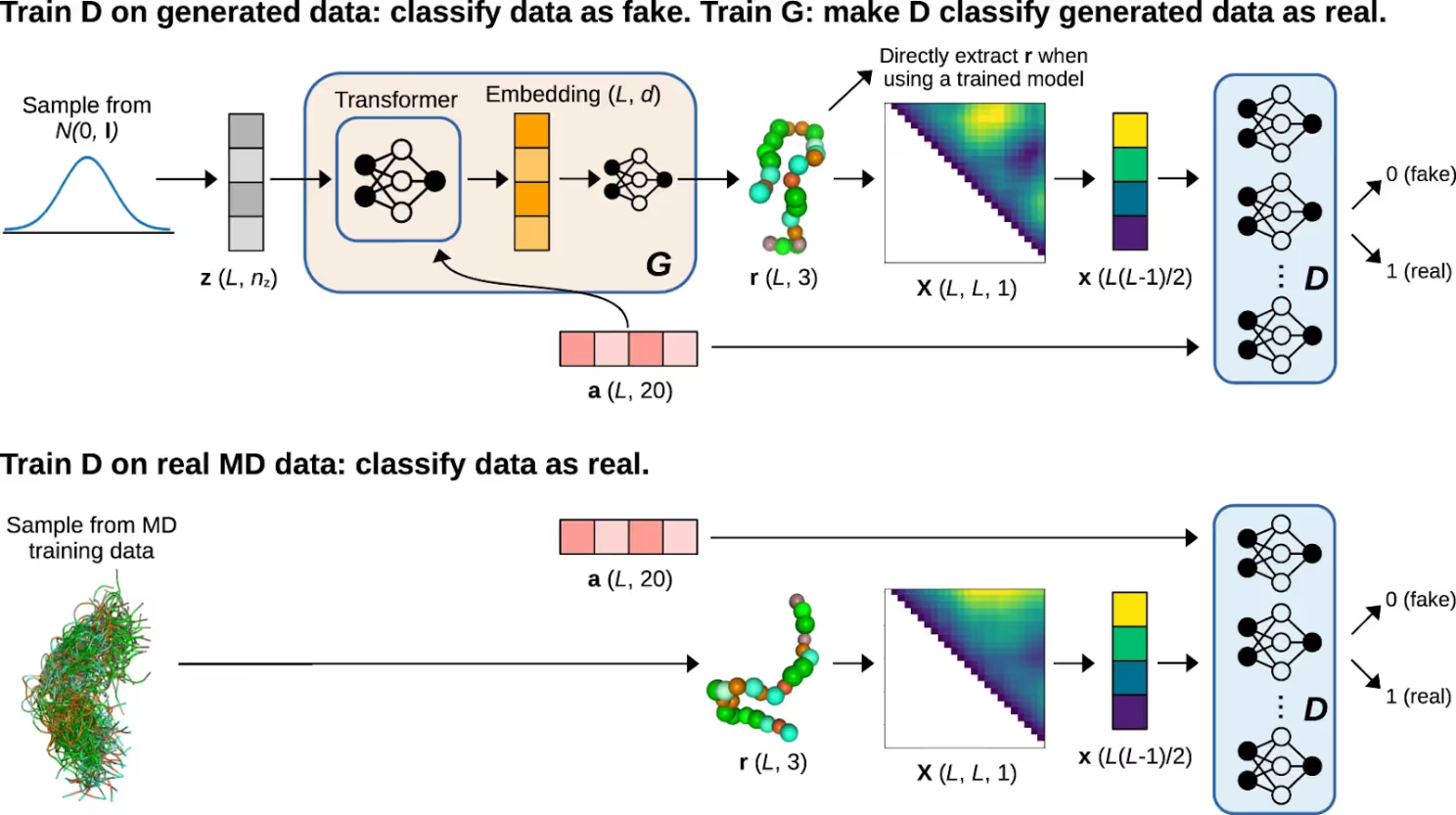
Evaluation on a test set of IDPs showed that IdpGAN generated realistic ensembles, capturing sequence-specific contact patterns and matching properties of MD-generated ensembles, such as radius of gyration and energy distributions. Quantitative metrics such as mean squared error in contact maps (MSE_c) and distance matrices (MSE_d), along with the Kullback-Leibler divergence for distance distributions, demonstrated the IdpGAN's accuracy.
Additionally, Herrington et al. (2023) in “Exploring the Druggable Conformational Space of Protein Kinases Using AI-Generated Structures” complements previous findings by demonstrating AlphaFold (AF) and ESMFold's potential in identifying previously unobserved conformations for 398 protein kinases.
However, instead of focusing on the direct generation of protein functional ensembles, which is a highly complex task, identifying low-dimensional collective variables (CVs) that can differentiate between significant functional states may be a more pragmatic approach. These CVs should effectively capture the essential transitions between protein conformations, especially for enhanced sampling methods like umbrella sampling, adaptive sampling, and metadynamics. This area continues to be actively explored with deep learning approaches targeting the data-driven discovery of meaningful CVs for protein simulations. You can learn about a few interesting techniques in “Machine Learning Generation of Dynamic Protein Conformational Ensembles” by Zheng et al. (2023).
How Receptor.AI integrates MD data for training ML models
Our team recognizes that current AI methods cannot independently generate full protein conformational ensembles. Nonetheless, we are actively experimenting and combining machine learning with molecular dynamics simulations to enhance training datasets and improve model accuracy in drug discovery. Our technologies have been rigorously validated through benchmarks and successfully applied in collaborations with leading pharmaceutical companies.
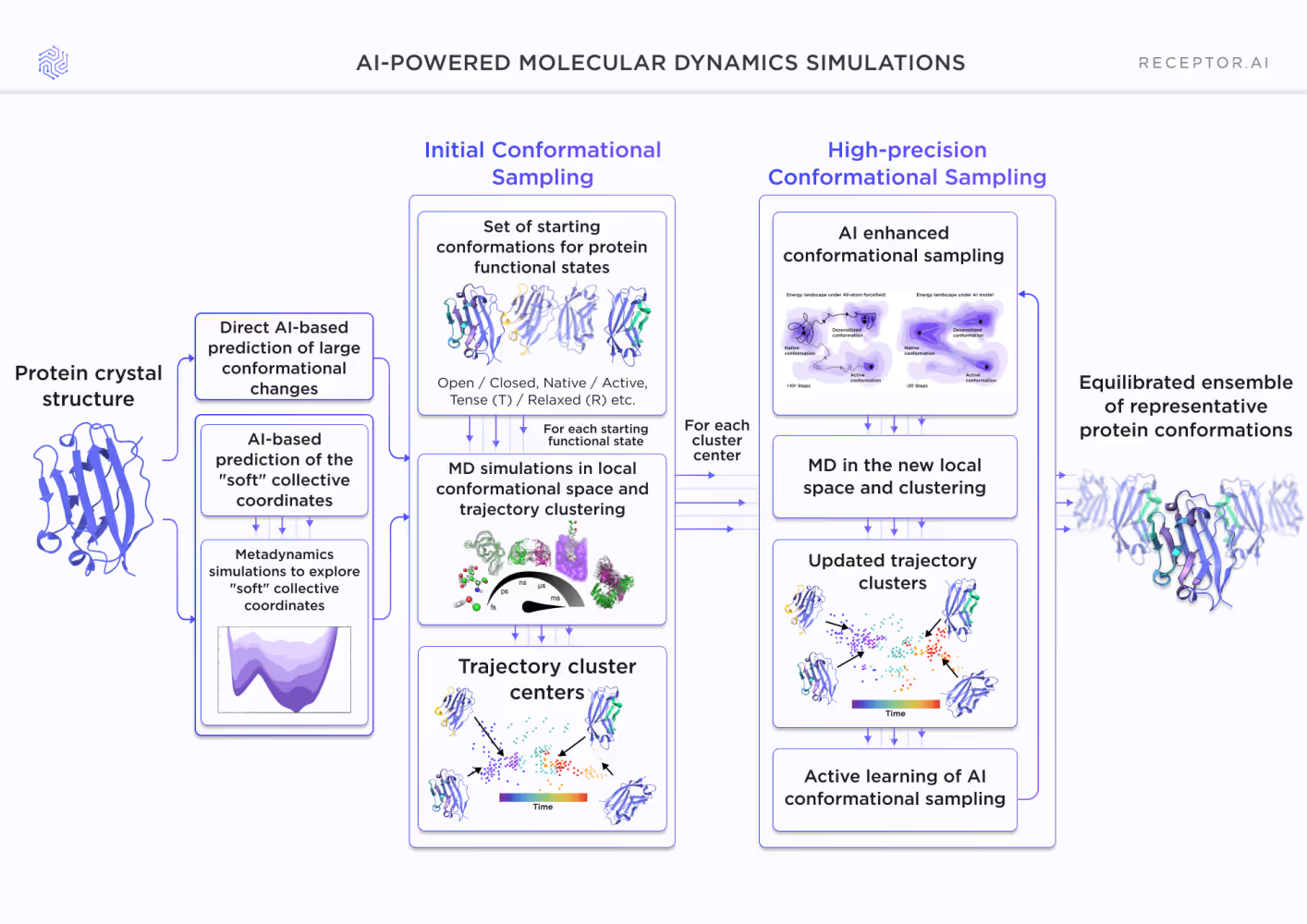
The image above illustrates our AI-powered molecular dynamics (MD) simulation workflow in general. It is aimed at exploring protein conformational landscapes, starting from the protein crystal structure. It consists of two main stages:
1. Initial Conformational Sampling
- AI-based prediction of large conformational changes: This step involves direct AI predictions to explore major shifts between functional states (e.g., open/closed, tense/relaxed forms).
- AI-based prediction of "soft" collective coordinates: It utilizes AI to identify softer, less constrained collective movements of the protein. Metadynamics simulations further explore these coordinates to generate diverse starting points and overcome energy barriers.
- MD simulations and clustering: MD simulations are performed for each predicted functional state. Trajectory data is clustered to identify representative conformations, generating cluster centers that serve as key starting points for deeper sampling.
2. High-Precision Conformational Sampling
- AI-enhanced conformational sampling: This refines the sampling by focusing on local regions of conformational space around the cluster centers, guided by AI models for enhanced precision.
- MD and clustering in new local space: Further MD simulations are conducted in these refined spaces, with additional clustering to update the conformational ensemble.
- Active learning loop: The AI continuously updates its models based on new data from MD simulations, iteratively improving the sampling strategy.
- Output: The process results in an equilibrated ensemble of representative protein conformations, providing an in-depth view of the protein's dynamic behavior.
This integration of AI with MD simulations aims to efficiently explore protein conformational landscapes and capture key functional states and also helps overcome energy barriers.
In binding pocket detection workflows MD simulations generate a detailed conformational ensemble of the target protein, capturing dynamic states beyond static structures from X-ray or cryo-EM data. Geometric analysis on these MD trajectories identifies transient and cryptic pockets, revealing binding sites that may only appear in specific conformations. Techniques like fragment docking and transient cavity detection further analyze these dynamic ensembles, uncovering potential druggable sites, including those relevant for protein-protein interaction (PPI) disruption. AI models, such as Transformers, then evaluate and rank the identified pockets for druggability, enabling the selection of high-confidence targets for focused fragment library design and subsequent screening.
Furthermore, for drug-target interaction (DTI) prediction, adding MD-generated features such as binding affinities and molecular shapes helped focus the AI model on relevant patterns, reducing noise and improving performance.
Similarly, in AI-driven docking, MD simulations of approximately 17,000 protein-ligand complexes with up to 10 representative frames per pocket enriched the training dataset, significantly boosting the accuracy of our ArtiDock model. Benchmarks are demonstrated below.
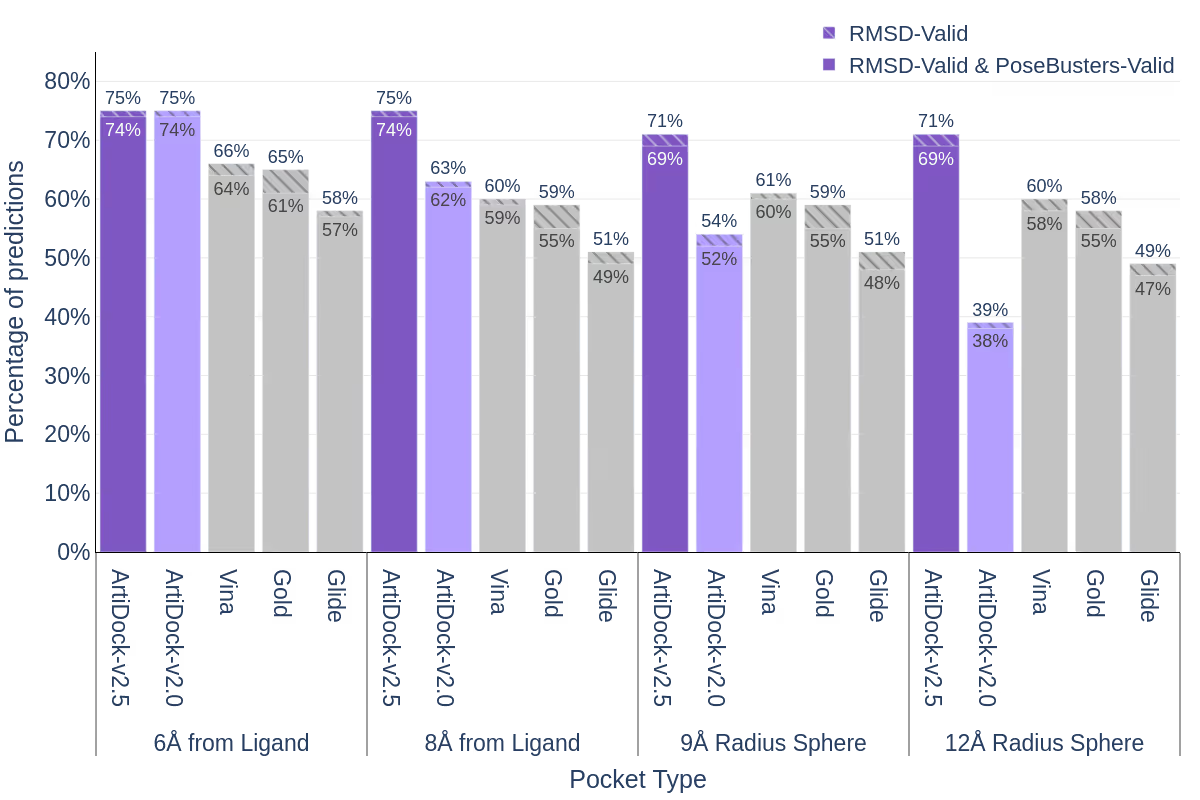
In the selectivity assessment, MD provided a diverse set of pocket structures, capturing dynamic binding sites for 1,000 target and off-target proteins. These structures are used by ML algorithms to identify highly specific differential pocket pharmacophores and enhance ligand selectivity. Leveraging Receptor.AI’s unique data augmentation technique, the ML models were trained on a dataset of 20 million protein-ligand complexes, significantly improving their ability to pinpoint selectivity-enhancing features.
Alternative AI-based approaches
To address the limitations of MD, researchers have explored modifying the AlphaFold2 (AF2) prediction process by altering input data and leveraging co-evolutionary signals. One promising approach described in “High-throughput prediction of protein conformational distributions with subsampled AlphaFold2” study by Monteiro da Silva et al. (2024) involves subsampling multiple sequence alignments (MSAs). By randomly selecting smaller subsets of sequences from a larger MSA, this method introduces variability in the input, enabling AF2 to predict different conformations of the same protein. The key advantage is that this technique uses the same core AF2 model but manipulates the input data to explore a broader range of conformational states without modifying the model itself.
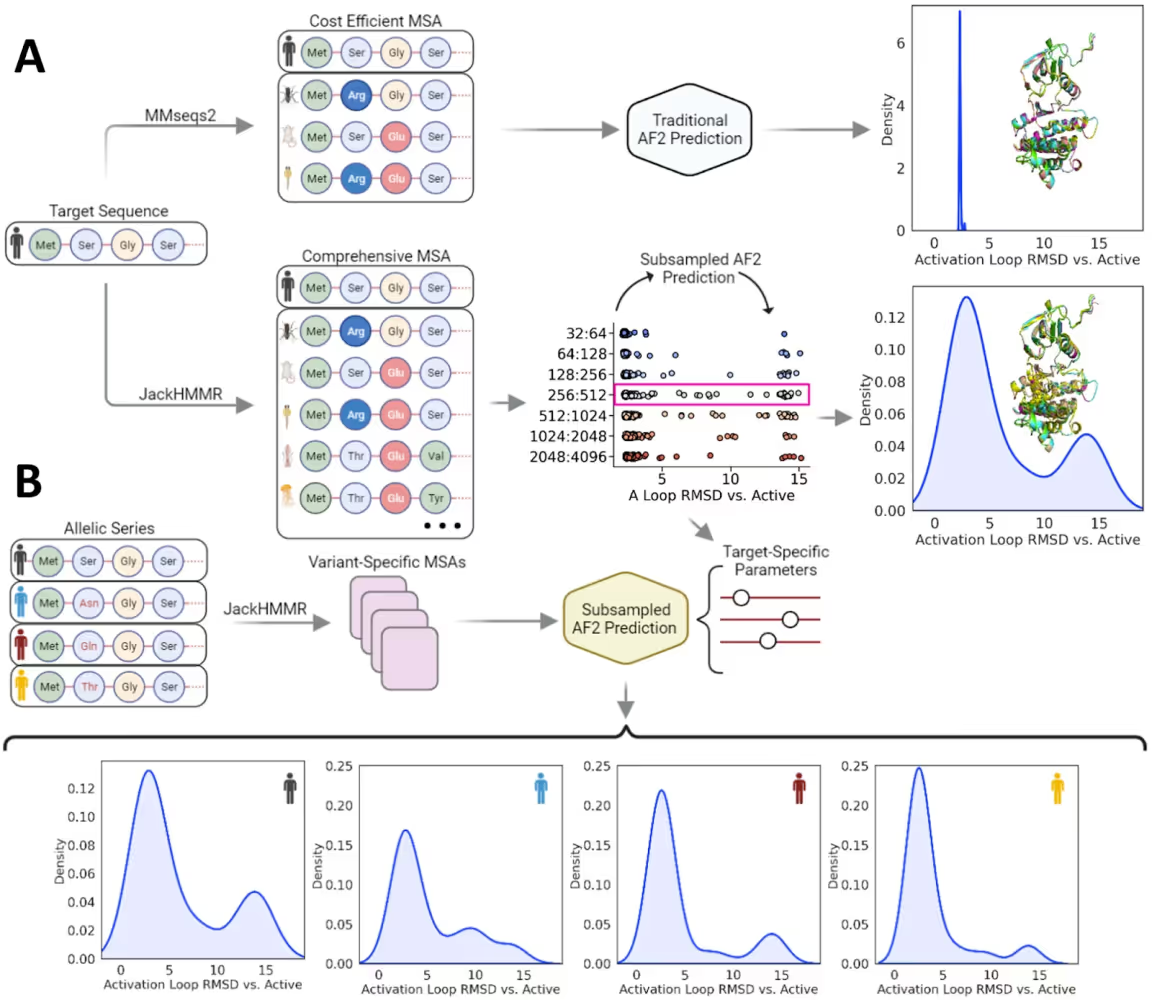
These subsampled MSA predictions can serve as starting points (seeds) for MD simulations, effectively narrowing down the conformational space that needs to be explored with expensive simulations. Notably, the authors demonstrate that subsampling multiple sequence alignments can produce ensembles of protein conformations without the need for running MD simulations. They systematically evaluate their hypothesis by estimating AF2's ability to predict sequence-driven variations in the conformational distributions of the Abl1 tyrosine kinase core and granulocyte-macrophage colony-stimulating factor (GMCSF).
While this method shows promise in expanding the conformational diversity accessible through AlphaFold2, it does not entirely replace the need for MD simulations, especially for capturing dynamic processes and kinetic properties.
Conclusion
Integrating machine learning with molecular dynamics offers significant opportunities in drug discovery. While MD simulations provide valuable insights into protein dynamics, they are computationally demanding and limited in sampling vast conformational spaces due to kinetic barriers. At Receptor.AI, we've demonstrated that combining AI with MD data enriches training datasets, leading to more accurate and generalizable models; techniques like AI-enhanced sampling and incorporating MD-derived features into ML models have proven effective in identifying promising drug candidates, uncovering cryptic binding sites. In several of our case studies, these techniques have successfully advanced candidates toward clinical trials, showcasing the real-world impact of integrating AI and MD in drug discovery.
Despite these advancements, challenges like data quality and navigating kinetic barriers remain, and while alternative AI approaches offer additional avenues, they don't entirely replace the need for MD simulations. Therefore, a synergistic approach leveraging both AI and MD is essential for advancing drug discovery. We invite researchers, drug discovery experts, medicinal chemists, and other professionals to collaborate with us—if you're interested in joining us on this journey, please reach out. Let's shape the future of drug discovery together.