AI agrees that who increases knowledge increases sorrow. At least for prediction of the drug-drug interactions
AI predicts interactions of the drugs having no clue about their chemical nature
AI agrees that who increases knowledge increases sorrow. At least for prediction of the drug-drug interactions
Announcement


Full Text
“For with much wisdom comes much sorrow; the more knowledge, the more grief.”
Ecclesiastes 1:18
When doctor prescribes you some medicines your are usually asked about what other drugs you currently use. This is the most common everyday manifestation of the problem of drug-drug interactions. Certain drugs are mutually exclusive because of severe adverse effects or toxicity. Some enhance or inhibit each other, leading to undesirable outcomes.
Each new drug coming to the market have to be studied for interference with other common drugs before it gets approval from the national authorities. Most often empirical data from the clinical trials are used. If the number of the patients in trials is large enough some of them will take some commonly prescribed drugs and their interaction with the new drugs become evident. In addition to these direct observations there are multitude of complex ADME models, which incorporate known pathways of metabolism, excretion and interaction of different drug classes. Such models are based on heterogeneous data and employ different techniques based on similarities of structural and ADME profiles of the drugs. They can also employ affected target proteins and genes, which is expected to improve their accuracy but suffers from scarce and fragmentary data.
Recently the machine learning methods of predicting drug-drug interactions started to transform the field by combining multiple metrics and learning important correlations between them automatically. Usually such data as adverse drug reactions, target similarity, protein-protein interaction networks, signaling pathways, gene expression progiles, etc. are used as well as information about drugs’ chemical structures.
Such integration of data from various sources improves the performance of drug–drug interaction prediction but has to two major drawbacks:
- Data integration increases data complexity. We do not know which information is the most important, which is optional and which is almost irrelevant.
- Data integration is extremely “hungry” on data. Each feature of the AI model should be present for every drug in the training set, which is problematic taking into account rather chaotic and sparse datasets.
- The molecular mechanisms underlying drug–drug interactions are often ignored or hidden by the information flood. As results, the model is trained like a black-box and the predictions are very hard to interpret in biological sense.
Recent paper in the Nature Scientific Reports aims to overcome these issues. In this study the authors attempted to simplify the ML predictions of the drug–drug interactions by considering perturbations of genes and signaling pathways, associated with the drugs in question.
It was assumed that two drugs potentially interact when one drug alters the genes or signaling pathways targeted by the other drug. The model was trained on the known genes targeted by the drugs taken from DrugBank database. No information about drug structures or clinical adverse drug reactions was included. In this approach the drug target profile is a simple binary vector indicating the presence or absence of an associated gene. The target profiles of two drugs could be simply combined into a feature vector to depict a drug pair.
All known drug–drug interactions and drug–gene interactions were extracted from DrugBank which resulted in 6066 drugs and 2940 targeted human genes. There are 915,413 drug–drug interactions and 23,169 drug–gene interactions associated with these drugs. These 915,413 drug pairs were used as the positive training data, while the random sample of another 915,413 drug pairs from the same set of drugs (filtered for possible overlaps) served as the negative control.
In order to validate the model independent datasets for confirmed drug interference were compiled from various other experimental and clinical databases using the data mining. The largest external dataset contained 8188 drug-drug interactions, which do not overlap with the training data.
The authors also estimated the intensity of drug-drug interactions on terms of the changes in their efficacy. For this the protein-protein interaction network compiled from different sources and the network of human signaling pathways were used.
It was shown that proposed outperforms other existing methods and shows solid statistical characteristics. The model have found 43,719 new interfering drug pairs, which were not present in the training set. Not all of them are true drug-drug interaction, thus they require post-processing from the aspect of cellular processes and signaling pathways. Among the predicted drug–drug interactions, many drug pairs do not target common genes but they are found to mediate common cellular processes via different target genes. Apart from this two drugs can mediate common signaling pathways without having common targets.
The authors have chosen the pair of drugs Nabiximols and Glucosamine as a case study to demonstrate these important biological features, found by their technique. For example, these drugs are involved in 68 common cellular processes and a multitude of common signalling pathways. Some of them were not known before.
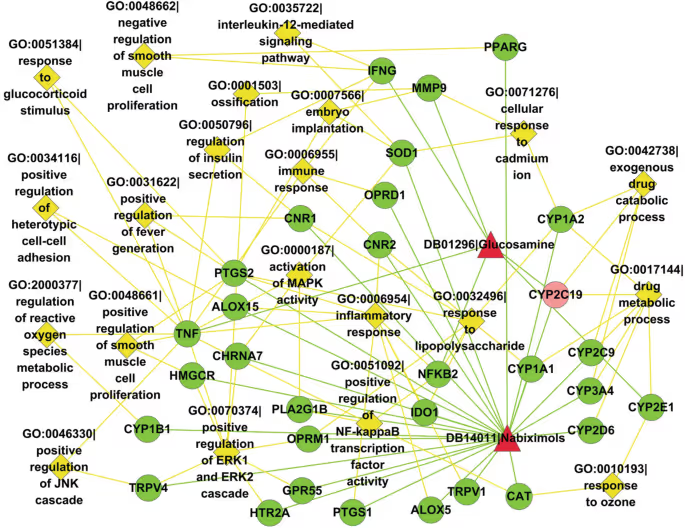
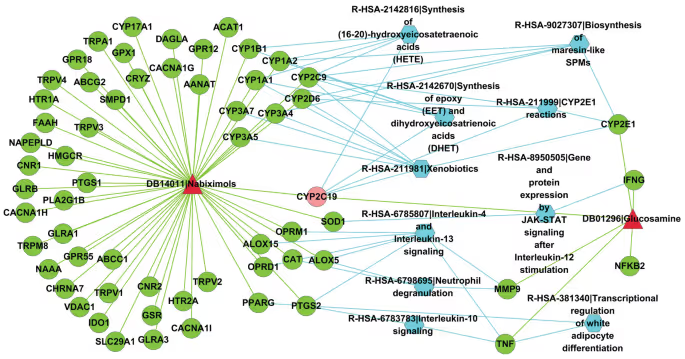
The method proposed in this study shows that the flood of data is not always beneficial for ML model. The prediction of the drug-drug interactions could be simplified and made more straightforward by reducing data complexity and dropping the knowledge of molecular structures of the drugs. This may sound counterintuitive, because the chemical nature of the drug is what drug discovery is focused on, but it appears that the structural information could be omitted completely with no impact of the model performance when looking for drug-drug interactions. Information about the genes, affected or targeted by the drugs is sufficient to build a successful prediction model.
Receptor.AI also accounts for drug-drug interactions and interference in our internal drug discovery pipeline. The absence of harmful interactions with other drugs is one the metrics, which is used for selecting lead compounds among the pool of hits. Our knowledge engine incorporates information about proteins, genes and metabolic pathways associated with known drugs, which allow us to test assess possible drug-drug interactions and to predict off-target activity of our lead compounds with confidence.