
Beyond Binding: Encoding Expert Strategy for Peptide Optimization
Announcement


Full Text
Most peptide projects we see today fail not because the peptide cannot bind. Between display technologies, fragment-based methods, and sequence-level AI, it is increasingly feasible to generate binders with nanomolar affinity to a target surface. The harder question is whether those binders can be turned into drugs: will they survive proteases, cross relevant barriers, and fit into a real formulation and IP strategy.
In other words, binding is necessary, but very far from sufficient. This is where our peptide work at Receptor.AI has been focused: not on inventing yet another scoring function in isolation, but on building an optimization agent within our PEPTOR™ platform that functions as a disciplined medicinal and computational chemist dedicated to a single peptide–protein complex.
From Medicinal Chemist Intuition to an Optimization Agent
Traditionally, peptide optimization is driven by a small group of experts who know the target class, have a mental library of successful motifs, and understand where permeability and stability tend to break. They look at structures, SAR tables, and assay data, and then propose a handful of mutations or cyclizations that “should work.”
We treated this process as something that could be formalized and encoded. The result is AI Orchestrator – an automated optimization framework within PEPTOR™ – that defines the compound design rules for each project:
- It ingests the target, desired peptide properties, and both open-source and proprietary data: peptide structures from literature and patents, homologous structural analysis, AI-derived SAR, and project-specific results.
- It chooses realistic starting points: PPI partner fragments, mRNA/phage display hits, or peptides generated directly in-pocket.
- It selects which parts of the peptide are allowed to move and how, based on key interactions and structural motifs that must be retained.
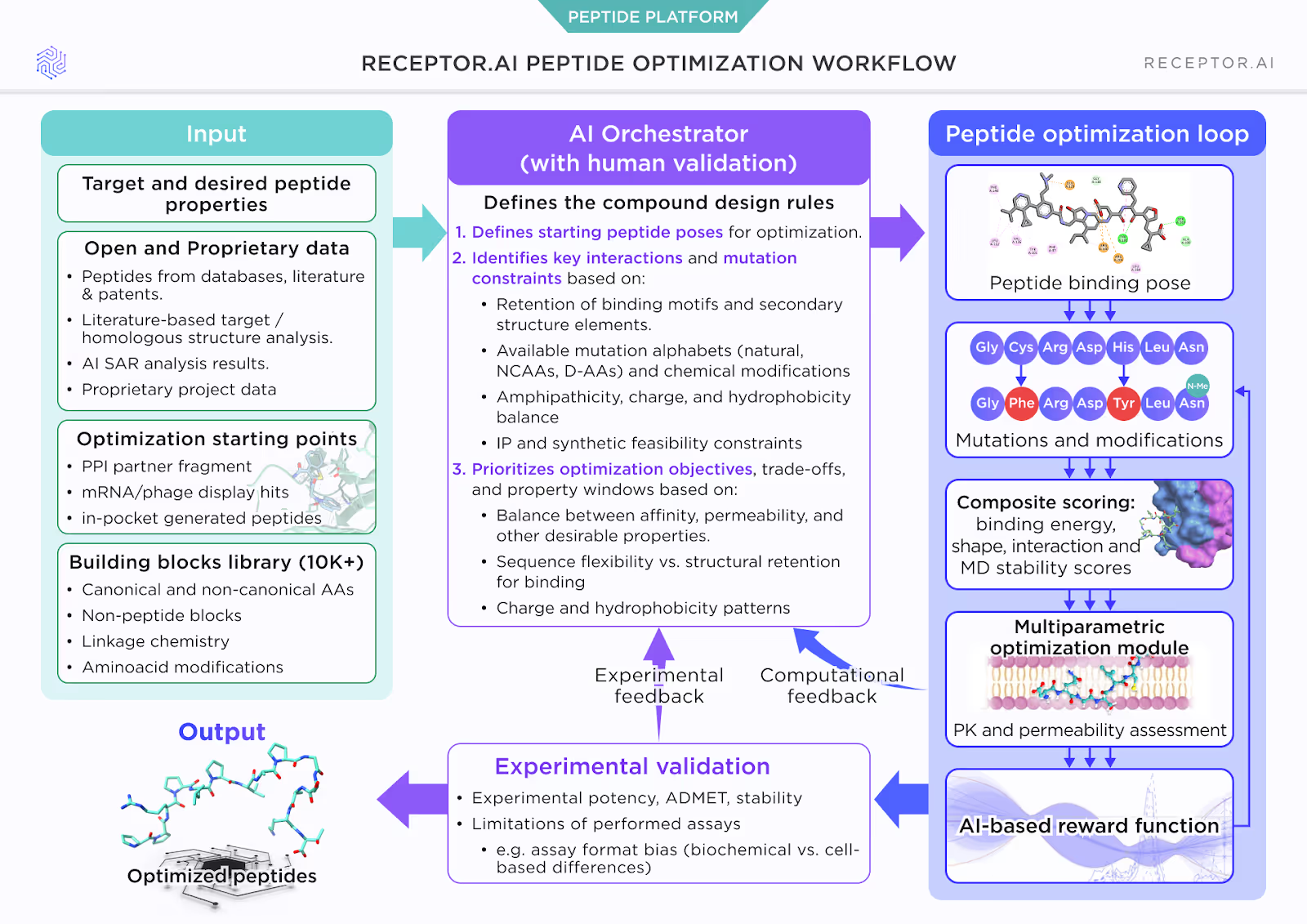
Under the hood, the Orchestrator is working with a very broad chemistry space: a building-block library of 10,000+ canonical and non-canonical amino acids, non-peptide blocks, linkage chemistries, and amino acid modifications.
The goal is not to generate arbitrary sequences, but to define a controlled search space where every proposal is chemically meaningful and aligned with the project’s constraints.
Binding Is Just One Term in the Objective
Once the design rules are set, PEPTOR™ runs a peptide optimization loop around a specific binding pose. For each candidate, it evaluates the peptide in complex with the target protein, not as an abstract sequence.
Conceptually, you can think of the loop as an optimization cycle guided by three layers of information:
- Physics-based metrics
- Interaction energies and shape complementarity in the binding pocket.
- Structural stability in the bound state.
- MD-based post-scoring to assess whether the peptide holds the desired conformation and whether the complex remains stable over relevant simulation timescales.
- Empirical, drug-likeness terms
- Amphipathicity, net charge, and hydrophobicity patterns that strongly influence permeability and solubility.
- Heuristics around proteolytic liability and chemical stability.
- IP and synthetic feasibility constraints: avoiding overly exotic motifs that are difficult to protect or manufacture at scale.
- PK and permeability assessment
- Dedicated modules provide estimates of passive permeability and related exposure parameters, feeding their outputs into the same reward function that scores binding and stability.
These components are combined into a composite score and then into an AI-based reward function that the optimizer tries to maximize. The system is explicitly multi-objective: it does not search for “the best binder,” but for candidates that sit in a property window balancing affinity, permeability, and other developability criteria.
In practice, this often means that the globally “best” binder is not selected. Slightly weaker binders with better simulated permeability and stability can rank higher, because they have a better chance of surviving the real pipeline.
Noncanonical Chemistry as a Core Design Dimension
A critical aspect of this setup is that PEPTOR™ is agnostic to how canonical the peptide is. The same optimization logic applies whether the sequence consists of natural amino acids, extensive noncanonical substitutions, backbone modifications such as N-methylation, or more complex linkers and caps.
This matters because many of the properties that kill promising peptides – permeability, metabolic stability, and formulation issues – are addressed most effectively in noncanonical space. If the design system cannot work natively with NCAAs, unusual linkers, and diverse cyclization strategies, it will push projects toward compromises that look good computationally but fail experimentally.
By integrating noncanonical building blocks directly into the searchable library and scoring them with the same physics-based and empirical framework, PEPTOR™ makes these modifications a default part of the exploration, not an afterthought added late in lead optimization.
What Changes for Real Projects
Putting this together, our peptide platform behaves like an automated medicinal/computational chemist working in a tight feedback loop with the lab:
- On the computational side, the AI Orchestrator proposes mutations and modifications to a specific peptide–protein complex, optimization loop evaluates them with physics-based and MD-informed metrics plus drug-likeness, and updates its internal representation of project-specific priorities.
- On the experimental side, potency, ADMET, and stability data feed back into the AI Orchestrator, correcting for assay limitations (for example, differences between biochemical and cell-based formats) and updating the reward function.
The role of human experts shifts accordingly. Chemists define objectives, constraints, and what success realistically means – across binding, exposure, and stability – rather than manually enumerating every plausible mutation. The Orchestrator handles the combinatorics and keeps the search grounded in physics and empirical knowledge.
For us, peptide design has become a clear example of where AI in drug discovery needs to go: away from single-task predictors and toward systems that encode expert strategy, reason over structures and properties in combination, and make multi-objective decisions under realistic constraints.
In future issues, I plan to share what we’re learning from these projects at Receptor.AI: when physics matters most, how noncanonical amino acids shift the optimization landscape, and what patterns we see across different target classes. For now, the main message is simple: in peptide therapeutics, binding is the easy part. The real work starts when you ask whether that binder can survive everything that comes after.