QuorumMap: AI-Guided Navigation of 10¹⁶-Scale Chemical Space
Announcement


Full Text
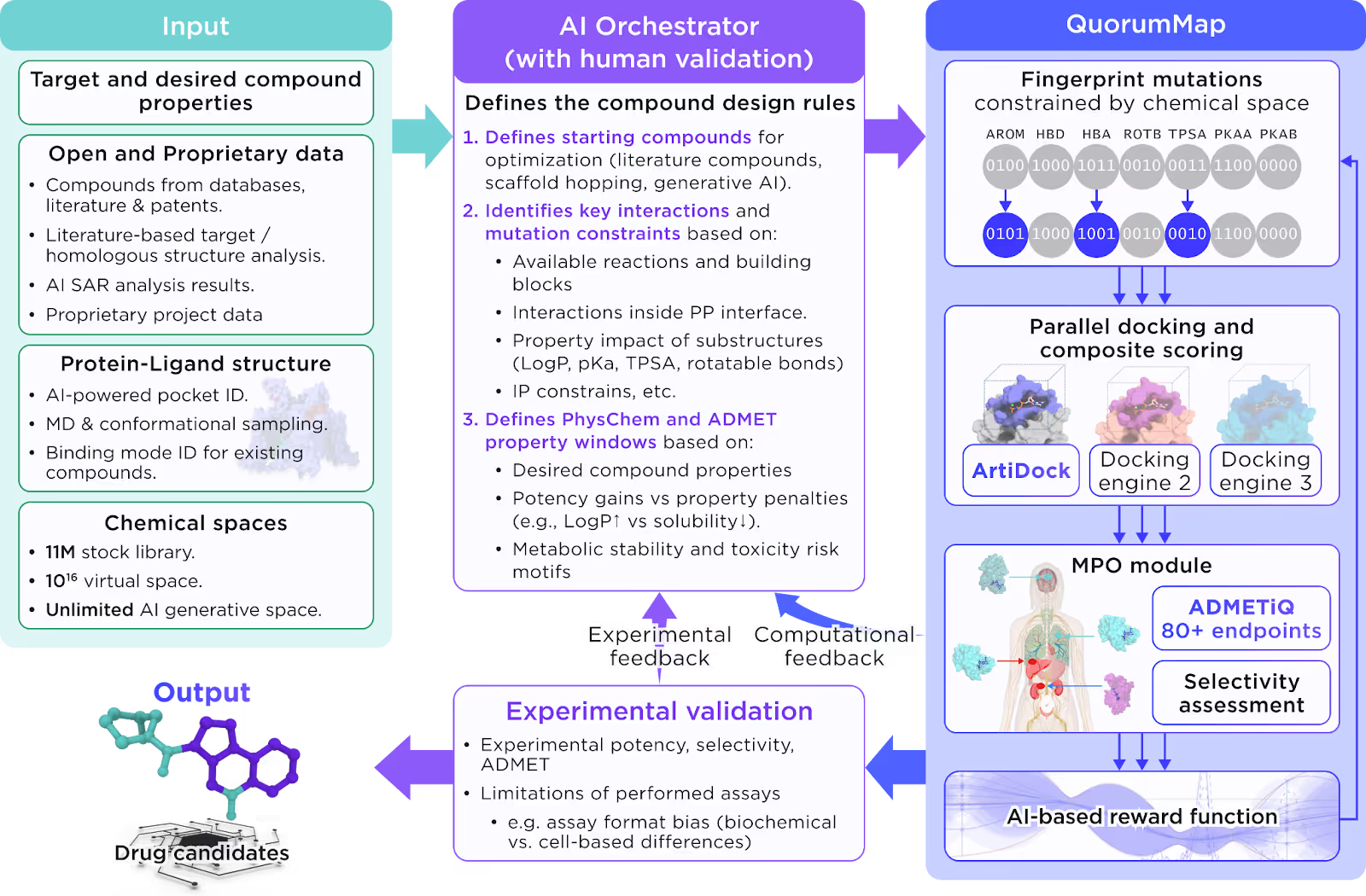
Receptor.AI has access to an infinite chemical space: over 10¹⁶ small molecules. No pharma team can dock, synthesize, or assay more than a tiny fraction of them. In Receptor.AI’s small-molecule platform, QuorumMap is the module that decides where to search. It uses AI to explore chemical space in small, information-rich batches, combines multiple docking engines and ADMET models, and continuously redirects the search based on experimental and computational feedback.
Navigating chemical space with uncertainty and diversity
QuorumMap does not try to sample the whole 10¹⁶-compound universe. It navigates from starting points through relevant chemotypes that meet project constraints (IP, target profile, route of administration, available chemistry).
Within each batch, compounds are ranked by an AI-based multi-parameter reward function that combines:
- docking and pose quality,
- ADMET and physicochemical properties,
- selectivity and drug-likeness,
- novelty/diversity and project-specific penalties.
Batch selection follows an uncertainty + diversity strategy:
- Uncertainty: prioritise compounds where the models are least confident to learn faster.
- Diversity: avoid near-duplicates and broaden chemotype coverage.
This inner optimisation loop runs autonomously: it repeatedly proposes, scores, and refines compounds based purely on the reward function, without invoking the AI orchestrator on every step.
Composite docking for more reliable ranking
Single-engine docking is noisy, whether physics-based or AI-based. Scores are sensitive to protein conformation, training bias, and pose artefacts. As we argued in our guest article “Rethinking docking reliability in drug discovery” on Scientist Live, combining complementary docking tools produces more stable and predictive results.
QuorumMap follows this principle: it runs parallel docking with several engines, including ArtiDock, keeps multiple poses per compound, and then re-scores them with Receptor.AI’s composite scoring function, leading to better enrichment of true binders and fewer wasted syntheses.
AI orchestration
The AI orchestrator operates at project-level checkpoints. At the start of a project it ingests:
- target and desired compound profile,
- internal and external small-molecule data,
- protein–ligand structures (including predicted pockets),
- stock, virtual, and generative chemical spaces.
From this it defines:
- initial starting points and chemotypes,
- allowed structural mutations and feasible reactions,
- PhysChem/ADMET windows and weights for the multi-parameter reward function.
After several optimization cycles and experimental or high-fidelity computational feedback (potency, selectivity, ADMET), the Orchestrator updates design rules, adjusts reward weights, and may open or close regions of chemical space based on what actually worked.
Conclusion
QuorumMap turns an unmanageable 10¹⁶-compound search problem into a guided optimization journey. By combining multi-parameter AI scoring, composite docking, and project-level orchestration that reacts to real experimental data, it helps pharma teams explore chemical space efficiently and stay focused on molecules that are not only strong binders, but also selective, developable, and aligned with the overall target product profile.